Translate this page into:
Pseudoxanthomatous or xanthelasmoid mastocytosis: Reporting a rare entity
Correspondence Address:
Preema Sinha
Department of Dermatology, Armed Forces Medical College, Pune - 411 040, Maharashtra
India
| How to cite this article: Sinha P, Sandhu S, Sen A, Sood A. Pseudoxanthomatous or xanthelasmoid mastocytosis: Reporting a rare entity. Indian J Dermatol Venereol Leprol 2020;86:382-385 |
Abstract
Mastocytosis is a disease characterized by abnormal and pathologic increase in mast cells in the cutaneous tissue and extracutaneous organs such as the bone marrow, liver, spleen, lymph node and gastrointestinal tract. Cutaneous mastocytosis comprises of four major clinical variants: solitary and multiple mastocytomas, urticaria pigmentosa, diffuse cutaneous mastocytosis and telangiectasia macularis eruptiva perstans. Cutaneous mastocytosis of the xanthelasmoid type is a rare variant of diffuse mastocytosis. It is clinically characterized by the typical yellowish hue and is accompanied histologically by mast cells infiltrating far into the lower dermis. Here we report one such rare case.
Introduction
Mastocytosis is a rare disease characterized by a primary pathological increase in the number of mast cells. These disorders present with varied clinical signs, symptoms and prognosis. The skin is the most commonly involved organ in all types of mastocytosis. Diffuse cutaneous mastocytosis is an extremely rare form of mastocytosis in which mast cells infiltrate the entire skin diffusely. Patients with this type of mastocytosis are at risk of systemic disease and severe complications but it often resolves spontaneously as in other types of childhood disease.[1],[2]
Case Report
An 11-year-old boy, born out of nonconsanguineous marriage presented with multiple raised pruritic yellowish to skin colored eruptions over the trunk and extremities, involving more than 70% of the body surface area for the past 2 years. Pruritus was severe enough to disturb his sleep. The child was delivered at an institution by vaginal route with an uneventful perinatal period and was exclusively breastfed with immunization up-to-date. There was a significant past history of recurrent episodes of vesiculo-bullous eruptions over the body since 11 months of age, which was managed at a government hospital till the age of 5 years. The vesiculo-bullous lesions were extremely pruritic and with symptomatic management had subsided by 5 years of age. The child was developmentally normal.
Examination revealed an averagely built boy at par with his age with no systemic abnormalities. Dermatological examination revealed multiple discrete to confluent yellowish papules and plaques predominantly over the trunk and also involving all four extremities with a rubbery consistency [Figure - 1]. The Darier's sign was negative.
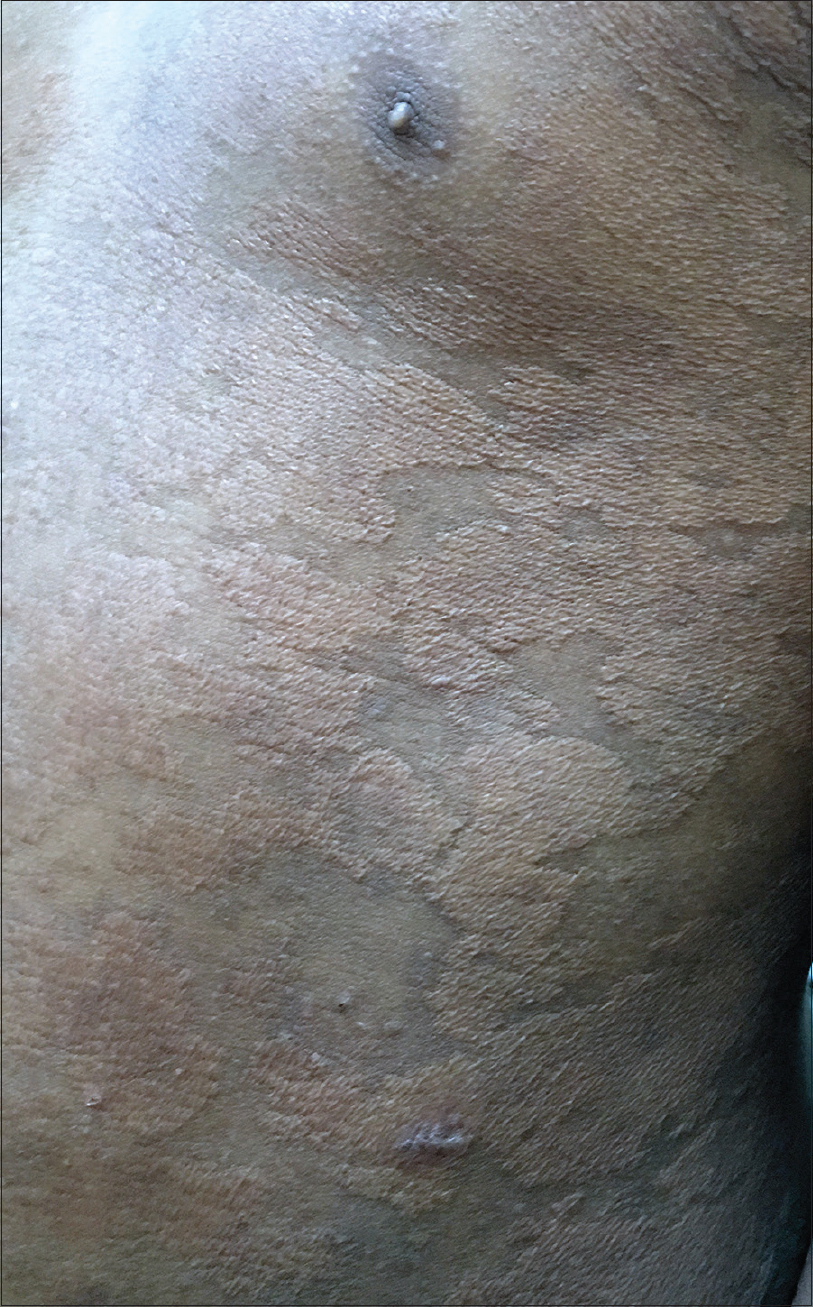 |
| Figure 1: Multiple discrete to confluent yellowish papules and plaques over trunk with a rubbery consistency |
Hematological, biochemical and urine analysis were within normal limits. Skin biopsy from the trunk revealed thinned out epidermis. Collection of mast cells in cords and nests was seen in both papillary and reticular dermis with many cells aggregated around the blood vessels [Figure - 2] and [Figure - 3]. These cells were monomorphic with abundant dense eosinophilic granular cytoplasm and dense round to eccentric nuclei. No involvement of deep reticular dermis was seen. Toluidine blue stain highlighted the dermal collection of mast cells [Figure - 4]. Immunohistochemistry was positive for CD 117, whereas negative for pan CK [Figure - 5]. Evaluation for any systemic involvement in the form of bone marrow studies was normal with 3% (mild) increase in mast cells. Serum tryptase level was 8 ng/ml. Based on the clinical, histopathological and immunohistochemical studies, he was diagnosed as a case of xanthelasmoid variant of diffuse cutaneous mastocytosis.
 |
| Figure 2: Collection of mast cells in cords and nests in dermis [hematoxylin and eosin (H and E), ×100] |
 |
| Figure 3: High power view of mast cells in dermis (H and E, ×400) |
 |
| Figure 4: Stain highlighted the metachromatic staining of mast cells (toluidine blue, ×400) |
 |
| Figure 5: Immunohistochemistry stain showed mast cells positive for CD 117 (immunohistochemistry, ×400) |
The child was managed symptomatically with oral antihistamines and topical emollients. The parents were counseled regarding the course and the outcome of the disease and avoidance of certain triggering factors including some drugs that may lead to mast cell degranulation and thereby flaring of the skin lesions. The patient has been under follow-up for the last 6 months with no fresh symptoms or signs.
Discussion
Mastocytosis is a disease characterized by abnormal and pathologic increase in mast cells in the cutaneous tissue and extracutaneous organs such as the bone marrow, liver, spleen, lymph node and gastrointestinal tract.[1] Mastocytosis is classified into three major clinical types: cutaneous, systemic and malignant forms. Cutaneous mastocytosis is a heterogeneous disorder that has the following major variants: urticaria pigmentosa, diffuse cutaneous mastocytosis, mastocytoma and telangiectasia macularis eruptiva perstans.[2]
Despite varied clinical features, the histological hallmark is the same for all types of cutaneous lesions with diffuse infiltration of mast cell in the dermis. The prognosis is good, but there is a risk of sudden mast cell degranulation due to any trigger and subsequent collapse. The most affected age group is <15 years with onset in infancy with a slight male to female predominance (1.5:1). Local and systemic symptoms in mastocytosis are caused by excessive production of mast cell- mediators such as histamine, leukotrienes, proteases and heparin.[3] The symptoms vary from patient to patient and include pruritus, dyspnea, flushing, blistering, bone pain, and features of gastrointestinal involvement in the form of vomiting, diarrhea and epigastric pain.[3],[4]
Diffuse cutaneous mastocytosis has two clinical variants: pseudoxanthomatous or xanthelasmoid and bullous type. Diffuse cutaneous mastocytosis is the rarest subtype, accounting for only 1.74% of all cases of cutaneous mastocytosis.[5] Diffuse cutaneous mastocytosis may present as a diffuse reddish-brown discoloration with a peau d'orange appearance affecting the entire surface of the skin, along with multiple yellowish or brownish doughy nodules resembling xanthomas or lesions of pseudoxanthoma elasticum, and is termed pseudoxanthomatous or xanthelasmoid mastocytosis. The exact prevalence of xanthelasmoid variant is not known but can vary from 8 to 25%.[6] Sometimes this variant has been accompanied by urticaria pigmentosa and juvenile xanthogranuloma.[7]
In patients with diffuse cutaneous mastocytosis, the first sign may be extensive bullae which may rupture, leaving erosions and crusts. The blisters may be of various sizes, initially contain clear fluid that may become hemorrhagic with time. Bullous lesions may occur in linear or grouped fashion and often develop on the trunk, scalp and extremities. These lesions typically resolve by 3–5 years of age without scarring. The blisters are believed to be caused by serine proteases that are released from the mast cells.[8]
Diffuse cutaneous mastocytosis can involve the entire skin with the central region and scalp most affected. The skin may be leathery and thickened. Hyperpigmentation may persist into adulthood and dermatographism may be prominent. Depending on the extent of the lesions and their severity, systemic symptoms can be present due to the large amount of mast cell mediators released locally and absorbed locally and systemically. Whole body flushing, pruritus, diarrhea, intestinal bleeding, hypotension, anemia, hypovolemic shock and deaths have been reported.[8] Visceral involvement with lymphadenopathy and hepatomegaly may be present. A rare complication includes pachydermia.[9] This form is particularly sensitive to psoralen-ultraviolet A with some reports of total or partial resolution after psoralen-ultraviolet A.
Pathogenesis of pediatric mastocytosis is different from the adult type. In adults there is a genetic inactivating mutation of the growth factor receptor c-kit. This mutation is absent in the pediatric cases which may explain the transient nature of the disease in them in comparison to adults.[9] However, children with progressive mastocytosis may express the same mutation as adults. Overall childhood mastocytosis has a good prognosis, but sometimes it can be progressive and fatal.
A study by Kettelhut et al. revealed that in most cases cutaneous mastocytosis in children does not involve internal organs which precludes the need for routine prognostic bone marrow biopsies.[10] Serum tryptase was significantly elevated only in children with systemic disease.[11]
Thus, cutaneous mastocytosis is a heterogeneous disease where neither the extent or the nature of skin lesions can predict the presence of systemic involvement. A better understanding of the pathogenesis would go a long way in understanding the disease.
One of the major concerns about diffuse cutaneous mastocytosis in pediatric patients is its association with systemic symptoms, such as debilitating diarrhea, gastrointestinal manifestations, shock and hypotension because of the much higher concentration of mast cells. However, in our patient, no systemic symptoms were observed during the hospital stay and inpatient visits.[6] Hence this case is being reported because of its rarity.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the parent has given the consent for images and other clinical information to be reported in the journal. The parent understands that names and initials will not be published and due efforts will be made to conceal patient identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Weedon D. Cutaneous infiltrates-non-lymphoid. In: Skin Pathology. 2nd ed. London: Churchill Livingstone; 2002. p. 1057-93.
[Google Scholar]
|
| 2. |
Husak R, Blume-Peytavi U, Pfrommer C, Geilen CC, Goerdt S, Orfanos CE. Nodular and bullous cutaneous mastocytosis of the xanthelasmoid type: Case report. Br J Dermatol 2001;144:355-8.
[Google Scholar]
|
| 3. |
Nayak S, Acharjya B, Devi B, Behera SK. Bullous mastocytosis. Indian J Dermatol 2007;52:201-3.
[Google Scholar]
|
| 4. |
Hartmann K, Henz BM. Mastocytosis: Recent advances in defining the disease. Br J Dermatol 2001;144:682-95.
[Google Scholar]
|
| 5. |
Fernandez AT, Campoamor LN, Mora LE, Zambrano AZ. Diagnosis, management and classification of pediatric mastocytosis. A study of 172 cases. Actas Dermosifiliogr 1998;89:461-76.
[Google Scholar]
|
| 6. |
Nabavi NS, Nejad MH, Feli S, Bakhshoodeh B, Layegh P. Adult onset of xanthelasmoid mastocytosis: Report of a rare entity. Indian J Dermatol 2016;61:468.
[Google Scholar]
|
| 7. |
De Villez RL, Limmer BL. Juvenile xanthogranuloma and urticaria pigmentosa. Arch Dermatol 1975;111:365-6.
[Google Scholar]
|
| 8. |
Hartmann K, Metcalfe DD. Pediatric mastocytosis. Hematol Oncol Clin North Am 2000;14:625-40.
[Google Scholar]
|
| 9. |
Walker T, von Komorowski G, Scheurlen W, Dorn-Beineke A, Back W, Bayerl C. Neonatal mastocytosis with pachydermic bullous skin without c-kit 816 mutation. Dermatology 2006;212:70-2.
[Google Scholar]
|
| 10. |
Kettelhut BV, Parker RI, Travis WD, Metcalfe DD. Hematopathology of the bone marrow in pediatric cutaneous mastocytosis. A study of 17 patients. Am J Clin Pathol 1989;91:558-62.
[Google Scholar]
|
| 11. |
Brockow K, Akin C, Huber M, Metcalfe DD. Assessment of the extent of cutaneous involvement in children and adults with mastocytosis: Relationship to symptomatology, tryptase levels, and bone marrow pathology. J Am Acad Dermatol 2003;48:508-16.
[Google Scholar]
|
Fulltext Views
3,775
PDF downloads
1,162





![[Figure - 1]](#fig_ijdvl_2020_86_4_382_252394_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2020_86_4_382_252394_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2020_86_4_382_252394_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2020_86_4_382_252394_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2020_86_4_382_252394_f5.jpg){kind=link}