Translate this page into:
Multiple, neonatal, self-healing, cutaneous glomuvenous malformations
Correspondence Address:
Alberto Conde-Taboada
Department of Dermatology, Hospital Clinico San Carlos, C/Prof. Martin Lagos, s/n, Madrid 28040
Spain
| How to cite this article: Conde-Taboada A, Campos L, Cuccolini L, López-Bran E. Multiple, neonatal, self-healing, cutaneous glomuvenous malformations. Indian J Dermatol Venereol Leprol 2017;83:226-228 |
Sir,
Glomuvenous malformations, formerly known as glomangiomas, arise from the neuromyoarterial elements of the glomus body and are histologically characterized by the presence of modified smooth muscle cells (glomus cells).[1],[2],[3] These benign tumors are identified by the presence of large and irregular vascular spaces covered by a thin coat of endothelial cells, further surrounded by a layer of glomus cells with a prominent central nucleus.[2],[4]
A 4-month-old girl was brought to our clinic with hard, purplish macular lesions on the back and on the upper and lower extremities, all of which had appeared progressively since birth [Figure - 1] and [Figure - 2]. None of the patient's relatives had similar lesions. A biopsy was taken from one of the macules. Histological examination revealed irregular, dilated vascular channels surrounded by cuboidal cells with a central nucleus and eosinophilic cytoplasm (identified as glomus cells) [Figure - 3]. A diagnosis of multiple cutaneous glomangiomas was made. It was decided to keep the patient under observation. At the next consultation, 3 months later, physical examination revealed that only two glomangiomas remained. The parents asserted that the child had been subjected to no procedure nor had she undergone any treatment. One year later, a complete skin examination revealed all the lesions to have disappeared [Figure - 4].
 |
| Figure 1: Two glomangiomas on the back (arrows) |
 |
| Figure 2: Blue lesion on the thigh (arrow) |
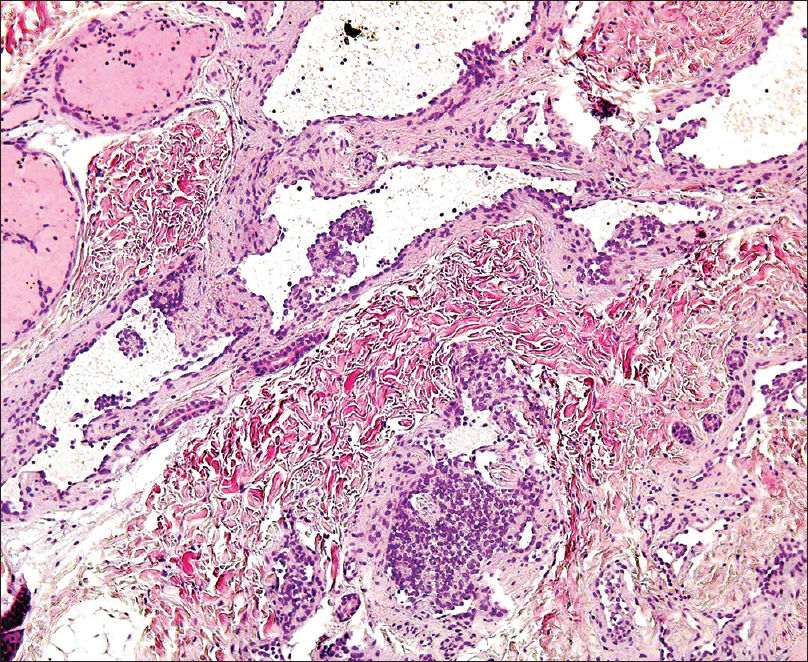 |
| Figure 3: Dilated vascular channels surrounded by glomus cells (H and E, ×400) |
 |
| Figure 4: The back of the patient showing resolution of glomangiomas, one year later |
Glomuvenous malformations appear at an early age as red-bluish papules or nodules; they are generally painless although some may be painful when palpated.[2] They can be classified into three variants: single glomus tumors (90% of all cases), multiple glomus tumors and jugular glomus tumors.[2],[4] Jugular glomus tumors are spherical conglomerations of cells and small blood vessels; they are therefore known as glomera.[5] An extremely rare congenital variant is also known that appears as papules which coalesce to form purplish plaques; these have been shown to grow with development.[2],[4]
Multiple glomuvenous malformations account for only 10% of all glomus tumors.[2] When they do appear, a family background is common (60% of cases); an autosomal dominant inheritance pattern with incomplete penetration has been described (70% at 5 years old; 100% at 30 years old).[1],[4] Glomuvenous malformations are associated with a mutation in the glomulin gene located on the 1p21-22 chromosome, the locus of which has been named VMGLOM.[1],[6] Congenital lesions are even rarer, with fewer than 20 cases described.[4] This is associated with an early postzygotic mutation (type 2 mosaic) originating from a loss of heterozygosity affecting a larger anatomical region.[4] Its association with type 1 neurofibromatosis has recently been discovered.[3] Differential diagnosis should include blue rubber bleb nevus syndrome; this presents with similar tumors (although with no glomus cells) and intestinal involvement.[2]
The present case represents an uncommon manifestation of glomangiomas since the patient had neither the plaques typical of the congenital type nor any associated family history. In addition, her tumors disappeared spontaneously. We were unable to find any similar previous reports. Indeed, one case of localized, partial regression of a plaque lesion which underwent several years of gradual involution has been described.[7] Segmental patterns of glomus tumors with unilateral, patchy presentations have also been reported in the literature. In those patients, new lesions appeared in their lifetime with no disappearance of the existing lesions.[8] Although rare, spontaneous remission has been reported in segmental genetic skin diseases such as epidermolysis bullosa or Kindler syndrome, each time involving changes in DNA correcting the germ line mutations.[9],[10] Although the present patient did not have segmental disease, reverting mutations may have played a role in inducing the spontaneous resolution.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.[11]
| 1. |
Boon LM, Brouillard P, Irrthum A, Karttunen L, Warman ML, Rudolph R, et al. A gene for inherited cutaneous venous anomalies (“glomangiomas”) localizes to chromosome 1p21-22. Am J Hum Genet 1999;65:125-33.
[Google Scholar]
|
| 2. |
Carvalho VO, Taniguchi K, Giraldi S, Bertogna J, Marinoni LP, Fillus JN, et al. Congenital plaquelike glomus tumor in a child. Pediatr Dermatol 2001;18:223-6.
[Google Scholar]
|
| 3. |
Brems H, Park C, Maertens O, Pemov A, Messiaen L, Upadhyaya M, et al. Glomus tumors in neurofibromatosis type 1: Genetic, functional, and clinical evidence of a novel association. Cancer Res 2009;69:7393-401.
[Google Scholar]
|
| 4. |
Allombert-Blaise CJ, Batard ML, Ségard M, Martin de Lassalle E, Brevière GM, Piette F. Type 2 segmental manifestation of congenital multiple glomangiomas. Dermatology 2003;206:321-5.
[Google Scholar]
|
| 5. |
De Smet LSciot RLegius EJ Med Genet. De Smet L, Sciot R, Legius E. Multifocal glomus tumours of the fingers in two patients with neurofibromatosis type 1. J Med Genet 2002;39:e45.
[Google Scholar]
|
| 6. |
VascularAnomaliesClassification: Recommendations from the International Society for the Study ofVascularAnomalies. Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, et al. Vascular Anomalies Classification: Recommendations from the International Society for the Study of Vascular Anomalies. Pediatrics 2015;136:e203-14.
[Google Scholar]
|
| 7. |
Kato N, Kumakiri M, Ohkawara A. Localized form of multiple glomus tumors: Report of the first case showing partial involution. J Dermatol 1990;17:423-8.
[Google Scholar]
|
| 8. |
Flórez A, Peteiro C, Sánchez-Aguilar D, Fernández-Redondo V, Pereiro Ferreirós M, Toribio J. Three cases of type 2 segmental manifestation of multiple glomus tumors: Association with linear multiple trichilemmal cysts in a patient. Dermatology 2000;200:75-7.
[Google Scholar]
|
| 9. |
Pasmooij AM, Pas HH, Bolling MC, Jonkman MF. Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting second-site mutations in LAMB3. J Clin Invest 2007;117:1240-8.
[Google Scholar]
|
| 10. |
Kiritsi D, He Y, Pasmooij AM, Onder M, Happle R, Jonkman MF, et al. Revertant mosaicism in a human skin fragility disorder results from slipped mispairing and mitotic recombination. J Clin Invest 2012;122:1742-6.
[Google Scholar]
|
| 11. |
Tolar J, McGrath JA, Xia L, Riddle MJ, Less CJ, Eide C, Keine DR, et al. Patient-specific naturally gene-reverted induced pluripotent stem cells in recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2014;134:1246-54.
[Google Scholar]
|
Fulltext Views
1,764
PDF downloads
925





![[Figure - 1]](#fig_ijdvl_2017_83_2_226_196319_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2017_83_2_226_196319_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2017_83_2_226_196319_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2017_83_2_226_196319_f4.jpg){kind=link}