Translate this page into:
Rothmund - Thomson syndrome with bronchiectasis: An uncommon phenotype?
2 Department of Pathology, Dr. R. P. Government Medical College, Kangra, (Tanda), Himachal Pradesh, India
Correspondence Address:
Vikram K Mahajan
Department of Dermatology, Venereology and Leprosy, Dr. R. P. Government Medical College, Kangra, (Tanda) - 176 001, Himachal Pradesh
India
| How to cite this article: Mahajan VK, Sharma V, Chauhan PS, Mehta KS, Raina R. Rothmund - Thomson syndrome with bronchiectasis: An uncommon phenotype?. Indian J Dermatol Venereol Leprol 2015;81:190-192 |
Sir,
We describe a case of Rothmund-Thomson syndrome (RTS; syn, poikiloderma congenitale) with bronchiectasis, a rarely reported association. [1] A 14-year-old boy had generalized poikiloderma since early childhood. He was the only child of non-consanguineous parentage and no one else in the family was similarly affected. Since 2 months of age, he would develop facial erythema and edema after breif sun exposure resolving in 3-4 days with hyperpigmentation. Generalized poikiloderma, of covered and uncovered skin, had manifested over the years [Figure - 1] and [Figure - 2]. He was reportedly treated for rickets (possibly radial hypoplasia), splenomegaly and anemia and had received blood transfusions at 5 years of age. He also experienced episodes of fever, cough with copious expectoration, and dyspnea since the age of six. He measured 136 cm (50 th percentile 164.1 cm, 3 rd centile 148.5 cm) in height and weighed 27 kg (50 th centile 51.2 kg, 3 rd centile 37.1 kg), suggestive of World Health Organization (WHO) grade I growth retardation (height deficit 83%, weight deficit 52.7%). He appropriately followed verbal commands and his mental age was 14 years on the Denver Development Screening Test. He had mild splenomegaly, left chronic suppurative otitis media, and crepts and rhonchi suggestive of obstructive lung disease. Ophthalmic, dental and neurological examinations revealed no abnormalities and his external genitalia were normal. Investigations revealed microcytic-hypochromic anemia with hemoglobin 11 g%, leucopenia with total leukocyte counts 3600/cmm and normal levels of serum immunoglobulins IgA, IgG and IgM. Bone marrow cytology showed normal maturation and mild plasmacytosis. A skin biopsy showed features of poikiloderma [Figure - 3]. The tuberculin test was <10 mm and pulmonary function tests showed features of obstructive lung disease. His chest X-rays revealed right middle lobe consolidation and reticular shadows in both lung fields. The skeletal survey was normal and his bone age was consistent with his chronological age. Computed tomography (CT) of chest revealed ground glass haziness in left upper, right middle and lower lobes, patchy consolidation and tubular bronchiectasis in right middle lobe, and mediastinal lymphadenopathy with focal calcification [Figure - 4]. Sputum grew Streptococcus pneumoniae on aerobic culture and showed no acid-fast bacilli in smears. His parents could not afford genetic studies. Genetic counseling, treatment for respiratory complications, vaccination against pneumococcal infection, photo protection/topical sunscreens, and long-term follow up were advised.
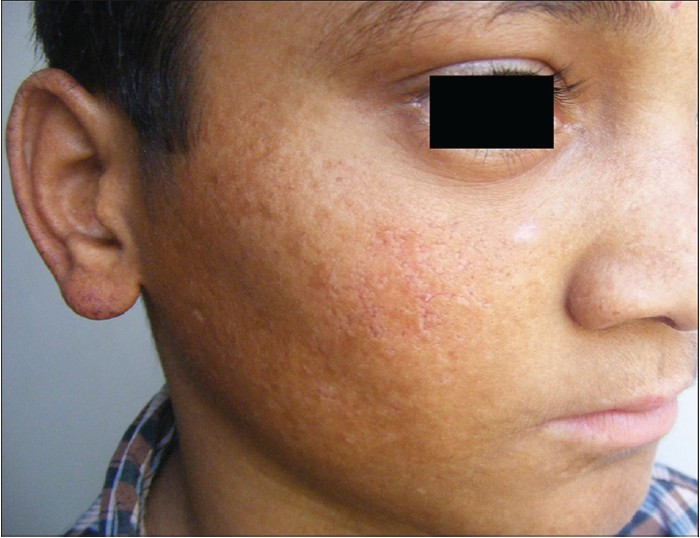 |
| Figure 1: Poikiloderma with reticulate erythema, hypo- and hyperpigmentation, atrophy, and telangectasiae over cheeks and pinnae especially on helix. Note normal eyebrows, eyelashes, and scalp hair |
 |
| Figure 2: Poikilodermatous skin over back and upper extremities |
 |
| Figure 3: Focal epidermal flattening, hydropic degeneration of basal layer, proliferation of blood vessels, inflammatory infiltrate comprising lymphocytes, histiocytes and a few melanophages in perivascular and periappendageal location suggestive of poikiloderma (H and E ×40) |
 |
| Figure 4: CT chest shows (a) tubular focal bronchiectasis, (b) patchy consolidation of right middle lobe, (c) ground glass haziness of right middle and lower lobes and (d) mediastinal lymphadenopathy |
Rothmund -Thomson syndrome is a rare autosomal recessive geno-photodermatosis affecting all races and both genders. This genomic instability syndrome results from defective deoxyribonucleic acid (DNA) damage repair to which an enhanced predisposition of these patients for skin cancers has been ascribed. Although the condition is attributed to homozygous or compound heterozygous (frameshift/missense) mutations in the RECQL4 DNA helicase gene mapped to chromosome 8q 24.3, this is detectable only in 60-65% cases. [2] Moreover, RECQL4 mutations have been identified in Baller-Gerold syndrome and RAPADILINO syndrome as well. These have many similar features; the difference between the two being presence of poikiloderma in the former and absence of it in the latter. [3]
Children with Rothmund-Thomson syndrome are usually born after normal gestation and consanguinity is not a constant feature. It presents in early infancy with photosensitivity manifesting as mild to intense erythema, edema and blistering after brief sun exposure that tends to decrease with age but may persist until adulthood. More typical poikiloderma over light exposed skin, face (cheeks, malar area, helices of ears) and extremities develop at 3-6 months or as late as 2 years of life and becomes generalized with advancing age. [4] Other heterogeneous features include short stature, sparse scalp hair, sparse/absent eyelashes and/or eyebrows, ophthalmic abnormalities (juvenile cataract, keratoconus, exophthalmos, glaucoma, prominent Schwalbe′s lines, absence of mesodermal layer of iris, tilted optic discs, tempo-retinal degeneration, pigment deposits over cornea and conjunctiva), skeletal abnormalities (frontal bossing, saddle nose, short stubby fingers, and congenital radial ray defects), premature aging, and a predisposition to malignancies (osteosarcoma, Hodgkin′s disease, myelodysplasias, acute myeloid leukemia) or skin cancers (squamous cell carcinoma, basal cell carcinoma, malignant fibrous histiocytoma). [4] Rare associated abnormalities include osteogenesis imperfecta, aminoaciduria, myelodysplastic syndromes, sensorineural deafness, defective dentition (microdontia, early caries), and hypogonadism in nearly 30% cases. [4] Physical growth is frequently retarded and most patients have short stature (occasionally to an extent of dwarfism), delicate limbs, small hands, and bird-like skull. [5] Lower respiratory tract infection, gastrointestinal (esophageal/pyloric stenosis, annular pancreas, chronic emesis, diarrhea) and hematological signs (leukopenia, microcytic hypochromic anemia) have been reported infrequently. [4] Despite such extreme clinical heterogeneity, the diagnosis remains largely clinical. Our patient had characteristic pokiloderma, growth retardation, anemia and possibly radial hypoplasia suggestive of Rothmund Thomson syndrome and also exhibited bronchiectasis and suppurative otitis media. Bronchiectasis remains an extremely rare manifestation and its pathogenesis is speculated to be due to: (i) vicious cycle of obstruction, recurrent infection, airway injury/inflammation and remodeling, (ii) impaired local host defenses for recurrent infections due to defective DNA helicase repair akin to cases of photosensitive trichothiodystrophy, (iii) chronic aspiration from swallowing, or (iv) an idiopathic event. [1] Isolation of S. pneumoniae from sputum in our patient and absence of recurrence after adequate treatment/vaccination supports the first two hypotheses. Although it remains speculative in the absence of gene mutation studies, it is a possibility that our case represents a new phenotype. The association of leucopenia and recurrent infections in some patients (including our patient) also suggests the possible existence of another phenotype with immunodeficiency. At least one patient of Rothmund Thomson syndrome with two unique genetic alterations in RECQL4 (IVS16-2A>T and IVS2+27_51del25) and combined immunodeficiency with T - B + NK - phenotype agammaglobulinemia has been described. [6] However, whether such cases are new phenotypes or are fortuitous associations remains conjectural due to the paucity of reported cases.
| 1. |
Reix P, Derelle J, Levrey-Hadden H, Plauchu H, Bellon G. Bronchiectasis in two pediatric patients of Rothmund- Thomson syndrome. Paediatr Int 2007;49:118-20.
[Google Scholar]
|
| 2. |
Kitao S, Shimarmoto A, Goto M, Miller RW, Smithson WA, Lindor NM, et al. Mutations in RECQL4 cause a subset of cases of Rothmund- Thomson syndrome. Nat Genet 1999;22:82-4.
[Google Scholar]
|
| 3. |
Irvine AD, Mellerio JE. Genetics and genodermatoses: Rothmund- Thomson syndrome. In: Burns T, Breathnach S, Cox N, Griffiths C, editors. Rook's textbook of dermatology. 8 th edn. West Sussex, UK: Wiley Blackwell Publishing Ltd.; 2010. p. 15.80-2.
th edn. West Sussex, UK: Wiley Blackwell Publishing Ltd.; 2010. p. 15.80-2.'>[Google Scholar]
|
| 4. |
Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis 2010;5:2.
[Google Scholar]
|
| 5. |
Sharma NL, Mahajan VK. Rothmund-Thomson syndrome. Indian J Dermatol 2003;48:110-2.
[Google Scholar]
|
| 6. |
Broom MA, Wang LL, Otta SK, Knutsen AP, Siegfried E, Batanian JR, et al. Successful umbilical cord blood stem cell transplantation in a patient with Rothmund-Thomson syndrome and combined immunodeficiency. Clin Genet 2006;69:337-43.
[Google Scholar]
|
Fulltext Views
1,596
PDF downloads
898





![[Figure - 1]](#fig_ijdvl_2015_81_2_190_152297_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_2_190_152297_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_2_190_152297_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2015_81_2_190_152297_f4.jpg){kind=link}