Translate this page into:
Designing a research protocol in clinical dermatology: Common errors and how to avoid them
Correspondence Address:
Saumya Panda
18-D/11, Anupama Housing Complex, VIP Road, Kolkata - 700 052, West Bengal
India
| How to cite this article: Panda S. Designing a research protocol in clinical dermatology: Common errors and how to avoid them. Indian J Dermatol Venereol Leprol 2015;81:115-123 |
"Dermatology is no longer a backwater for evidence-based medicine (EBM), although it still has a long way to go"
Hywel C. Williams, ′Where do we go from here?′ in Evidence-Based Dermatology (2014)
Indian dermatology is currently passing through an interesting phase. One of its positive features is the upswing in the quantity and quality of clinical research that is being performed and published. At the same time, the scope of various sources of institutional funding has also widened for the dermatological researcher to make use of. In such a scenario, the prospective clinical researcher must have very clear ideas about designing and writing clinical protocols. Unfortunately, scrutiny of many such protocols submitted in the recent past as part of research grant applications in dermatology has revealed various lacunae, particularly in the areas of research design, methodology, and statistical analysis. In this article, our endeavor will be to point out the common pitfalls in protocol designing and writing by our clinical researchers and to suggest ways to prevent them.
The clinical research flow is, generally speaking, a unidirectional one according to the following schematic:
Idea →Design → Protocol → Analysis
The Idea
The research idea (question/problem/hypothesis) comes first and drives the study design. It should be original, simple, and logical. The researchability of a question or problem is judged by the F (feasibility) I (interestingness) N (novelty) E (ethicality) R (relevance) [FINER] criteria. Ultimately, this gets reflected in the protocol, which is conventionally judged by these criteria.
Example
Is 0.5% timolol maleate gel-forming solution effective and safe in the treatment of pyogenic granuloma?
The Title
The title of the protocol should reflect the idea in a nutshell and describe the type of study being undertaken.
Example
Effectiveness and safety of 0.5% timolol maleate long-acting aqueous solution in the treatment of pyogenic granuloma - a multicentric, randomized, double-blind, placebo-controlled pilot study
Example of an inadequate title
Efficacy of 0.5% timolol versus placebo in pyogenic granuloma.
The latter title does not give an idea whether the trial was an open one or a blinded one, randomized or not, whether side effects were compared, the precise formulation of timolol, etc. One of the common errors is mistaking ′effectiveness′ and ′efficacy′ as synonyms. While efficacy studies may be defined as a test of whether an intervention does more good than harm when administered under optimum conditions, effectiveness studies are defined as a test where the intervention is administered under real-life conditions. [1]
The major differences between the two kinds of trials are highlighted in [Table - 1]. [2] Interventions are often less effective in clinical settings than in the laboratory. Similarly, interventions may be less successful in effectiveness trials than in efficacy trials.

The Introduction
This is a very important, if less emphasized, section that requires utmost attention. It should cover the scientific background, the statement of the problem, and the a priori hypothesis. The former should be presented in a concise manner based upon an extensive and integrative review of the current literature. The introduction of a protocol may be viewed as analogous to the perspective of a drawing. Just as the perspective lends the appearance of depth or distance in a drawing, the introduction should serve to describe the depth of the problem on one hand, and give an idea what to expect from the study on the other.
Hypothesis may be defined as the explanation of the relationship between two or more variables. It is not a haphazard guess ("Does eating brinjal worsen psoriasis?"); it is not imagination run riot either ("Is melasma related to neurofibroma?"); nor is it meant to simply test a fortuitous statistical association ("a correlation between owning cats and being struck by lightning"). A correct formulation of hypothesis is, again, based on a proper understanding of the problem at hand and a critical analysis of current literature.
The Objective
Specific objectives that flow from the hypotheses, along with planned subgroup analyses, wherever appropriate, must be stated at the outset.
Example
Primary objective: To assess the effectiveness and safety of topical timolol maleate in the treatment of pyogenic granuloma
Secondary objective: To evaluate the patient′s subjective assessment of the effect of treatment.
The Methodology
This is the most important aspect of the protocol that helps conduct high quality research and obtain valid inferences based upon the research question. A complete clinical research protocol includes:
- Study design;
- Precise definition of the disease or problem;
- Completely defined pre-specified primary and secondary outcome measures, including how and when these will be assessed;
- Clear description of variables;
- Well-defined inclusion and exclusion criteria;
- Efficacy and safety parameters;
- Whenever applicable, stopping guidelines and parameters of interim analyses;
- Sample size calculation;
- Randomization details;
- Plan of statistical analysis;
- Detailed description of interventions;
- A chronogram of research flow (Gantt Chart);
- Informed consent document;
- Clinical research form;
- Details of budget; and
- References. [3]
In short, the researcher should clearly state the PICO (Patient, Intervention, Comparison, and Outcome) elements of the research question in this section.
The study design
The study design defines the objectives and endpoints of the study, the type and manner of data collection and the strategy of data analysis. The different types of clinical studies have been summarized in [Figure - 1]. The gold standard design for a clinical therapeutic trial is parallel group, double-blind, randomized and controlled. This is usually referred to as randomized controlled trial (RCT). [4] The suitability of various study designs vis-à-vis different types of research questions are outlined in [Table - 2].
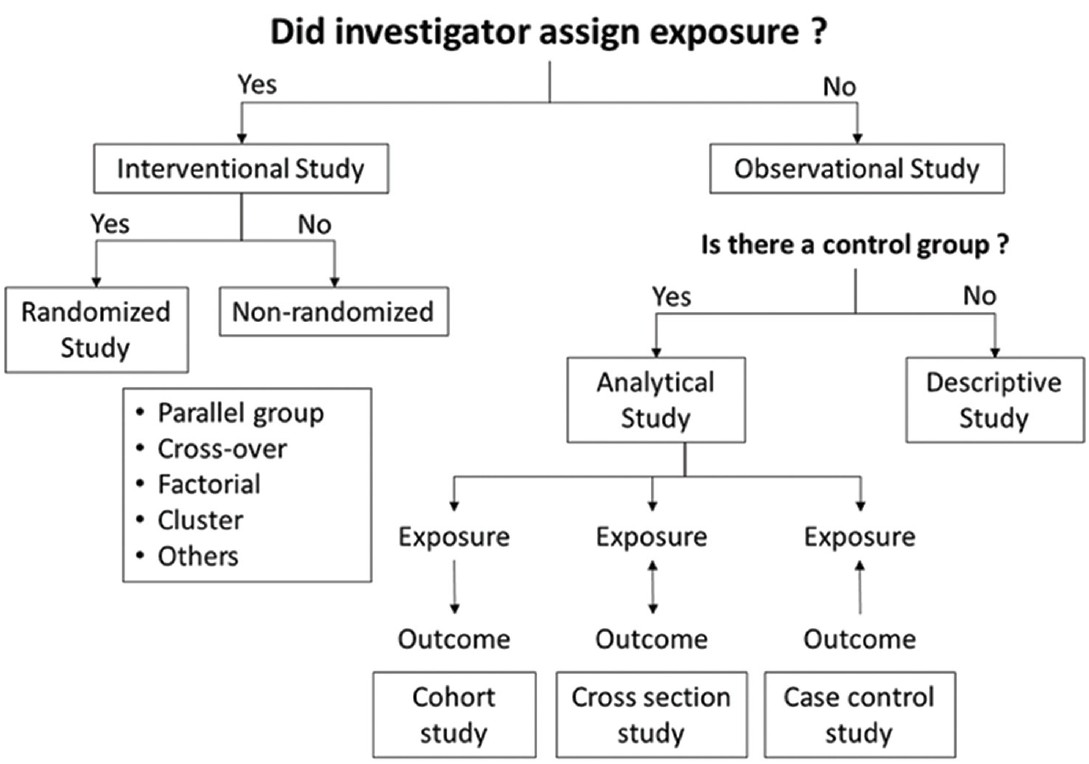 |
| Figure 1: Types of study |

Clinical studies are experiments, but these are not conducted in laboratories but in controlled real-life settings on human subjects with some disease. So, designing a study involves many pragmatic considerations aside pure methodology. Thus, factors to consider when selecting a study design are: objectives of the study, time frame, treatment duration, carry over effects, cost and logistics, patient convenience, statistical considerations, sample size, etc.
In a conventional clinical trial, the goal is to demonstrate superiority of one intervention over another (superiority trial). However, a superiority trial that fails to demonstrate superiority cannot be automatically inferred to have demonstrated either non-inferiority or equivalence. A trial has to be designed prospectively so as to demonstrate conclusively either of the latter outcomes. So, along with superiority trials, we have two other kinds of clinical trials: non-inferiority (no worse than) trials and equivalence (as good as) trials.
Certain truisms regarding study designs should always be remembered: a study design has to be tailored to objectives. The same question may be answered by different designs. The optimum design has to be based upon manpower, budgetary allocation, infrastructure, and clinical material that may be commanded by the researchers. Finally, no design is perfect, and there is no design to provide a perfect answer to all research questions relevant to a particular problem.
Example
An interventional, randomized, parallel-group, multicentric, placebo-controlled, prospective, evaluator-blinded therapeutic trial including 40 patients, aged 2-65 years, with clinically diagnosed pyogenic granuloma over skin, to evaluate efficacy and safety of 0.5% w/v timolol maleate long-acting solution (2 drops twice daily applied to the lesion) in 20 patients versus glycerine 2% solution twice daily (2 drops applied to the lesion) in 20 patients for a period of 6 weeks.
Bias, controls, blinding, randomization
The greatest enemy of a clinical trial is bias. The bias may arise from a number of sources in the trial, for example, from the patient to whom the treatment is being administered, from the clinical staff who administer the treatment, and the researcher (analyst) who interprets the results (data).
In order to reduce such bias, a number of strategies have been devised. One such is introducing controls in a study. In an uncontrolled study, there is no comparator treatment. On the other hand, a treatment may be compared with placebo (placebo-controlled); two different doses of the same drug may be compared (dose-response study); one treatment may be compared with another (active control); or, the control may be external (same treatment may be administered on two different subsets of the same disease, e.g. characterized by a key difference in histories with the same condition).
Another important strategy is blinding (masking) in clinical trials. This means concealing the exact identity of a treatment. This prevents patients, investigators, and assessors from getting influenced by the identity of the treatment. This also reduces the tendency for bias to enter into the evaluation of the trial results. Thus it reduces performance, assessment, reporting, and analysis bias.
In an open-label (non-blinded) study, everyone, including patients, is aware of the exact identity of the intervention. In a single-blind study, only the patient is unaware of the treatment. In a double-blind study, both the patient and the investigator (attending clinician) are unaware. In a triple-blind trial, the patient, the investigator as well as the analyst are all unaware of the exact identity of the intervention administered.
In an active controlled study, double dummy blinding is needed, when the investigational agent and the active control are dissimilar in appearance. In such situations, a placebo in the investigational drug arm of the study should resemble the active control, while the placebo in the active control arm should resemble the test drug.
Breaking the blind may be necessary to protect the patient in case of severe untoward events. Defined procedures for breaking the randomization code under such circumstances exist, which should be specified in the protocol beforehand. The mechanism should be such that the blind be broken for the individual patient, and not for the whole study.
In some studies, there could be ethical concerns about continuation of the study, either when the active treatment is so effective that the subjects in the placebo or no treatment arm may be deemed to be unfairly deprived of the same, or when the test intervention is so very ineffective that there is a high probability of serious harm or mortality in the study. In such studies, arrangement for interim analysis must be provided for in the protocol at predetermined time points, with a priori stopping boundaries or threshold values for interrupting the trial, according to various Bayesian statistical models.
Subjects should come from the same population, especially if cultural, ethnic, and socioeconomic diversity have the possibility to interfere with either the disease or the treatment or both. Age and gender are classical restrictions in homogenizing group responses. Therefore, they should be allocated in such a way that each participant has the same chance of being assigned to each group. [5] Randomization is the process by which subjects are allocated in a manner that ensures that each subject has an equal chance of receiving one or the other of the treatments being evaluated, thus minimizing selection bias. It is a systematic process, and is not the same as allocation at random.
Randomization circumvents the natural tendency of the researchers to select the perceived best treatment for their patients. Randomization also ensures that baseline factors, which may influence the observed differences in treatment outcome, get equally distributed between treatment groups. Thereby, randomization improves the chance of drawing a valid statistical inference regarding association between treatment and outcome.
Thus, non-random allocation methods, viz., convenience based, snowball, voluntary response, saturation, quota sampling, etc., and quasi-randomized techniques, viz., alternate allocation, according to date of birth, day of attendance at the clinic, etc., are to be avoided. Different types of randomization schedule according to the needs of the study, for example, simple, stratified, block or cluster randomization, may be adopted. The method used to generate the random allocation sequence (e.g. random number table, software), and details of any restriction (viz., blocking and block size) have to be specified. The mechanism used to implement the random allocation sequence (viz., sequentially numbered containers) should be specified, describing the steps taken to conceal the sequence until interventions are assigned. Ideally, it should be mentioned in the protocol as to who among the investigators would generate the random allocation sequence, who would enroll the participants, and who would assign the participants to interventions at each site.
Outcome measures for efficacy and safety
These should be objective and quantitative, and should be assessed at different time points. Correlation between the results at each time point reinforces the statistical validity of the effect. Confounders should be considered and assessed for adjustment of their effects.
Example
Primary outcome measures: Difference in color of pyogenic granuloma in the treatment group versus control group; physicians′ global assessment (on 4-point Likert scale) based on blind photographic evaluation; spontaneously reported adverse events or those elicited by the clinicians, and changes in laboratory values (complete blood count, liver function, renal function, blood sugar levels), all assessed at 2-weekly intervals for 6 weeks
Secondary outcome measures: Patients′ or parent′s subjective global assessment (on 4-point Likert scale) at 2-weekly intervals for 6 weeks.
Inclusion and exclusion criteria
Inclusion and exclusion criteria must be clearly spelt out at the outset. These should be comprehensive and categorical. It should be clearly understood that exclusion criteria does not include any of the parameters specified in the inclusion criteria, that is, these criteria are independent of each other.
Example
Inclusion criteria: Age 2-65 years, pyogenic granuloma only over skin surface, duration of lesion not more than 8 weeks, informed consent or assent
Exclusion criteria: Pregnant or lactating women; immunosuppressed condition due to disease and/or medication; systemic comorbidities (cardiac problems, bronchial asthma, chronic obstructive pulmonary disease, hypoglycemia); concurrent use of systemic β-blockers, calcium channel blockers, quinidine or any medication having significant interaction with β-blockers; and previous treatment for pyogenic granuloma.
Sample size calculation
This is one of the most misunderstood, yet fundamentally important, issues among clinicians and has to be addressed once study objectives have been set and the design has been finalized. Too small a sample means that there would be a failure to detect change following test intervention. A sample larger than necessary may also result in bad quality data. In either case, there would be ethical problems and wastage of resources. The researcher needs just enough samples to draw accurate inferences, which would be adequately powered.
Sample size determination is a statistical exercise based upon probability of errors in testing of hypothesis, power of the sample, and effect size. Type I or α error is wrongly rejecting the ′no effect′ (null) hypothesis, that is, when the chance of getting a false positive result occurs. Usually, size of type I error specified before data correction (α) is 0.05 or 0.01.
Type II error or β error is wrongly concluding that there is no effect, that is, when a chance of a false negative result occurs. Usually, β =0.2 (or 0.1). Statistical power (1-β) represents the probability of correctly rejecting the null hypothesis when it is false. α is reciprocal to 1-β. Usually, power is taken to be 80%. Ideally, it should be 90%. Decreasing type II error increases power, but it also increases type I error. Effect size (δ) is the smallest clinically important difference expected between means in the two groups, and is often expressed as a multiple or fraction of the standard deviation (SD). Thus, it is also referred to as the standardized difference.
Sample size formula for test of difference between two means:

where N = total sample size for two equal sized groups
σ1 = SD in group 1
σ2 = SD in group 2
δ = effect size
Zα = value corresponding to significance level
Zβ = value corresponding to power level
Sample size formula for test of difference between two proportions:

where p 1 = expected proportion of the outcome in group 1
p 2 = expected proportion of the outcome in group 2
Adjustments in this calculation are needed for dropouts, for unequal sized groups, for multiple outcomes, and if subgroup analysis is to be performed.
In order to adjust group size for attrition rate, the sample size (N) has to be multiplied by the correction factor 100/100-X, where X = dropout rate in percentage (%).
For unequal sized groups, adjusted sample size N= N × (1 + k) 2/ 4k, where k = ratio of larger to smaller group size.  is the number of subjects to be allotted to the smaller group and the rest to the larger group.
is the number of subjects to be allotted to the smaller group and the rest to the larger group.
Lehr′s formula is a quick calculation of sample size for power of 80% and a two-tailed significance level of 0.05. The required size of each of two equal groups is.  For power of 90%, the sample size becomes
For power of 90%, the sample size becomes  .
.
If standardized difference is small, this formula would tend to overestimate the sample size.
In practice, sample size is determined by either of these formulae, tables based on these formulae or statistical software.
Example:
- Assumptions
- α =0.05 (two-sided)
- Power (1 - β) =0.8
p 1 = 0.71 (proportion of resolution in the group treated with topical timolol, from the single published uncontrolled series of topical timolol in pediatric cases of pyogenic granuloma that showed resolution in five cases out of seven [5] ).
p 2 = 0.2 (proportion of resolution in the control group).

Estimated required sample sizes.
n 1 = 18
n 2 = 18
Assuming an attrition rate of 10%, sample size becomes 20 in each group. Thus, total sample size is 40.
Software and web resources for power and sample size calculation
There are numerous software, many of which can be freely downloaded, for power and sample size determination. Examples: PS (a free interactive program), PASS (Power Analysis and Sample Size Software; provides sample size tools for over 650 statistical test and confidence interval (CI) scenarios; a trial version can be downloaded for free), GFNx01Power, NQuery Advisor, Pc-Size, etc.
Also, there are several websites, many of which are hosted by various universities, which provide sample size calculators. Examples: David Schoenfeld′s Statistical Considerations for Clinical Trials and Scientific Experiments (Harvard), UCLA Calculator Service, Sample Size and Power - Survival Outcomes (Johns Hopkins School of Public Health), Rollin Brant′s Sample Size Calculators (University of Calgary), Simple Interactive Statistical Analysis, Russ Lenth′s Power and Sample Size (University of Iowa), etc.
Statistical analysis
Not only every researcher, but also every student and practitioner of clinical sciences, including dermatology, should have a basic knowledge of statistical methods and data analysis in order to be able to choose the optimal study design and interpret the data.
Data structure depends on the characteristics of the variables [Figure - 2]. Quantitative data always have a proportional scale among values, and can be either discrete (e.g. number of acne lesions) or continuous (e.g. age). Qualitative data can be either nominal (e.g. gender) or ordinal (e.g. Fitzpatrick′s phototypes I-VI). Variables can be either binary or dichotomous (male/female) or multinominal or polychotomous (homosexual/bisexual/heterosexual). The variables whose effects are observed on other variables are known as independent variables (e.g. risk factors). The latter kind of variables that change as a result of independent variables are known as dependent variables (i.e. outcome). Confounders are those variables that influence the relation between independent and dependent variables (e.g. the clinical effect of sunscreen used as part of a test intervention regimen in melasma). If the researcher fails to control or eliminate the confounder, it will damage the internal validity of an experiment.
 |
| Figure 2: Data types and classification of variables |
Numerical data can be either parametric or nonparametric. Whether the distribution of data in the underlying population is normal may be tested with a normal probability plot or normality tests, viz., Shapiro-Wilk or Kolmogorov-Smirnov test, etc. If the distribution is parametric (normal), data should be represented by mean and standard deviation. If not, median and interquartile ranges are generally used. Skewed data, however, may be transformed and subsequently analyzed with parametric tests. Assumptions of parametric tests require that observations within a group are independent, which samples have been drawn randomly from the population, and that samples have the same variance, that is, drawn from the same larger sample. The last assumption (homogeneity of variances) may be assessed through a test, for example, Levene′s test. Large samples (say n > 100) approximate a normal distribution and can almost always be analyzed with parametric tests.
The clinical research questions needing the help of appropriate statistical tests in order to derive valid inferences may be broadly classified into two categories: one asking whether there is a difference between two groups, and the other asking whether there is an association or agreement between two variables or assessments. [Table - 3] and [Table - 4] summarize these two categories of questions and the statistical tests needed to help answer these, respectively.


The study population may be analyzed either as intention-to-treat (ITT) or any of its modifications, or per protocol (PP). In any interventional trial, ITT population includes all subjects who have been recruited or randomized, and who may have subsequently dropped out, whereas PP includes only those who completed the study. Performing an ITT analysis seeks to eliminate attrition bias, and is often regarded as a major criterion by which the risk of bias of an RCT is assessed. However, it may be entirely appropriate to conduct a PP analysis alongside the ITT analysis, particularly in equivalence or noninferiority trials. [6]
Statistical significance is usually set as a two-tailed (nondirectional) P < 0.05, which means that the maximum probability of getting the observed outcome by chance is less than 5%. The null hypothesis (position of no change) is rejected if P < 0.05. Although P > 0.05 means insufficient evidence to demonstrate difference between groups, it should be remembered that it is not the same as no difference.
Representative samples are needed to draw inferences regarding populations. Confidence interval (CI) is a measure of the precision of the results obtained from sampling; 95% CI means that 95% of such intervals will contain the population value. In other words, it is the range of values within which it can be precisely said, with 95% confidence that the true population value lies. The two values that define the interval are called 95% confidence limits. The convention of using 95% CI is arbitrary.
Statistical modeling is done to find out a predictive mathematical relation between an outcome (dependent) variable and one or more explanatory (independent) variables. Examples: simple linear regression, logistic regression, Cox proportional hazards model, general linear models, and various multivariate techniques. Unpaired data are usually analyzed by multiple logistic regression (categorical dependent variable) or general linear models (quantitative dependent variable). Paired data are usually analyzed by generalized mixed models. [7] Discrete data in Poisson or negative binominal distribution are analyzed by generalized mixed or linear models. Adjustment of results by confounders is important to explore data and multivariate analysis is frequently used for that purpose.
The difference between two treatments is usually summarized as the difference between two means if the data is continuous (e.g. psoriasis area and severity index). For categorical data, where the outcome is binary (response or no response) there are a number of ways to summarize a treatment difference. The choice of summary method can affect how the treatment difference is interpreted. Relative risk (RR), relative risk reduction (RRR), absolute risk reduction (ARR), and number needed to treat (NNT) are some of the important measures of treatment difference. NNT, which measures the benefits of treating a population, is the reciprocal of ARR. It is a very intuitive way to understand the magnitude of benefit relative to baseline risk. It refers to the number of patients that would need to be treated in order to see one additional success in the new treatment in comparison with another treatment. Thus, a new treatment that results in clearing of psoriasis in 40% of patients, compared with 30% for the conventional treatment, translates into a risk difference of 40% - 30% =10% and an NNT of 10 (1/0.1). In other words, one needs to treat 10 patients to clear one additional patient. [8]
Ethical and regulatory requirements
The consent form should be written in a simple and lay language in vernacular and link languages. The objectives of the study, potential risks, benefits, and detailed procedures should be clearly described. The investigators′ contact numbers should be displayed in the consent form. The protocol must be submitted to the respective Institutional Ethics Committee (IEC) where the study would take place. In case of a clinical trial, the IEC must be registered in India with the Central Drugs Standard and Control Organization (CDSCO). In case of a new drug or an existing drug to be tested for a new indication even in academic trials have to be submitted to the office of the Drugs Controller General of India (DCGI). Finally, all clinical trials, following approval by the respective IECs, must be registered with our official clinical trials registry, that is, the Clinical Trials Registry - India (CTRI).
Here we have tried to address, in a nutshell the common inadequacies noticed among the protocols submitted by Indian dermatologists as part of grant applications. To summarize, the following suggestions/recommendations may be offered for qualitative improvement of our protocols:
- The title has to clearly and comprehensively reflect the kind of study being undertaken.
- The study must have novelty, particularly in the Indian context.
- Objectives of the study must be clearly and cogently stated.
- Inclusion and exclusion criteria must be explicit and complete.
- The design of the study must conform to the outcomes. For example, if safety is the primary outcome, a dose-ranging study has to be performed.
- Even in an open study, a control arm is desirable.
- Sample size calculation must be properly done a priori.
- A proper statistical analysis is mandatory, including multivariate analysis to tackle confounding variables, wherever appropriate.
- ITT analysis in all interventional studies, and in particular, RCTs should be the standard.
- Proper construction of a research timetable and an appropriate disaggregated budget are necessary parts of all proper protocols.
- A clinical research form and an informed consent document are mandatory.
- The current norms of following standards of ethics, and obtaining requisite permission from the regulatory bodies are a must.
- And last but not least, the protocol should be as free from grammatical and linguistic errors as possible.
Research today is a very highly skilled vocation that demands various skill sets in addition to academic passion and scientific curiosity. A model protocol is the reflection of design and sampling skills, observation and measurement skills, data management skills, analysis and interpretation skills, and writing and communication skills of the research team. So, build a team with these exceptional skills and, definitely, you will generate an exceptional protocol.
ACKNOWLEDGMENT
Examples have been provided from an existing protocol of a study currently underway. I acknowledge here the contributions of the investigators and co-authors of the protocol: Drs. Feroze Kaliyadan, Maninder Singh Setia, Sujay Khandpur, Sunil Dogra, Chitra Nayak, Nilay Kanti Das, Amrita Sil, and Avijit Hazra.
| 1. |
Flay BR. Efficacy and effectiveness trials (and other phases of research) in the development of health promotion programs. Prev Med 1986;15:451-74.
[Google Scholar]
|
| 2. |
Glasgow RE, Lichtenstein E, Marcus AC. Why don't we see more translation of health promotion research to practice? Rethinking the efficacy-to-effectiveness transition. Am J Public Health 2003;93:1261-7.
[Google Scholar]
|
| 3. |
Bagatin E, Miot HA. How to design and write a clinical research protocol in cosmetic dermatology. An Bras Dermatol 2013;88:69-75.
[Google Scholar]
|
| 4. |
Appel LJ. A primer on the design, conduct, and interpretation of clinical trials. Clin J Am Soc Nephrol 2006;1:1360-7.
[Google Scholar]
|
| 5. |
Wine Lee L, Goff KL, Lam JM, Low DW, Yan AC, Castelo-Soccio L. Tretament of pediatric pyogenic granulomas using β-adrenergic receptor antagonists. Pediatr Dermatol 2014;31:203-7.
[Google Scholar]
|
| 6. |
Schiller P, Burchardi N, Niestroj M, Kieser M. Quality of reporting of clinical noninferiority and equivalence randomised trials - update and extension. Trials 2012;13:214.
[Google Scholar]
|
| 7. |
Norman GR, Steiner DL. Biostatistics. The Bare Essentials, 3 rd ed. Shelton, Connecticut: People's Medical Publishing; 2008.
[Google Scholar]
|
| 8. |
Williams HC. Applying the evidence back to the patient. In: Williams HC, Bigby M, Herxheimer A, Naldi L, Rzany B, Dellavalle RP, et al., Editors. Evidence-Based Dermatology, 3 rd ed, Chichester, Sussex: John Wiley and Sons; 2014.
[Google Scholar]
|
Fulltext Views
3,204
PDF downloads
1,127





![[Table - 1]](#tbl_ijdvl_2015_81_2_115_152168_t7.jpg){kind=link}
![[Figure - 1]](#fig_ijdvl_2015_81_2_115_152168_f9.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2015_81_2_115_152168_t8.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_2_115_152168_f10.jpg){kind=link}
![[Table - 3]](#tbl_ijdvl_2015_81_2_115_152168_t11.jpg){kind=link}
![[Table - 4]](#tbl_ijdvl_2015_81_2_115_152168_t12.jpg){kind=link}