Translate this page into:
Trichorhinophalangeal syndrome type I - Clinical, microscopic, and molecular features
Correspondence Address:
Chil Hwan Oh
Department of Dermatology, Korea University Guro Hospital, Gurodong gil 97, Guro dong, Guro-gu, Seoul, 152 - 703
Korea
| How to cite this article: Jeon J, Kim JH, Oh CH. Trichorhinophalangeal syndrome type I - Clinical, microscopic, and molecular features. Indian J Dermatol Venereol Leprol 2014;80:54-57 |
Abstract
Trichorhinophalangeal syndrome type I (TRPS I) is an autosomal dominant malformation syndrome characterized by a triad of hair alteration, craniofacial and skeletal abnormalities. TRPS1 gene was first identified in 2000 and mapped on chromosome 8q23.3. A 39-year-old female patient with short stature (149 cm) visited for fine sparse and slow-growing hair with receded medio-occipital hairline of roughly triangular shape since infancy. A typical pear-shaped nose and elongated philtrum were noticeable. In addition, she reported deviation of middle phalanges, bilateral coxa varus in both hips and brachydactyly on bilateral fourth digits. Mutation analysis identified a transition of cytosine to thymine at position 1630 (exon 4), which results in amino acid change R544X and a premature stop of translation. There is no established treatment. But through careful evaluation of suspicious cases to identify potential mutation carriers, the patient can receive information about the disease and genetic counseling.Introduction
TRPS I, first described by Giedion in 1966, is an autosomal dominant genetic disorder characterized by the triad of hair alteration, craniofacial and skeletal abnormalities. [1] TRPS1 gene was first identified in 2000 and mapped on chromosome 8 that includes 7 exons and encodes a polypeptide of 1,281 amino acids. [2],[3]
Different forms sharing similar clinical features are TRPS II characterized by the presence of mental retardation, multiple cartilaginous exostoses or microcephaly in addition to the features of TRPS I and TRPS III, which has similar clinical features to TRPS I except that TRPS III presents more severe brachydactyly and growth retardation. [4],[5]
Case Report
A 39-year-old Korean female patient with short stature (149 cm) visited for fine sparse and slow-growing hair with receded medio-occipital hairline of roughly triangular shape since infancy. A receding fronto-temporal hairline was also observed and a typical rarefaction of lateral eyebrows, pear-shaped nose, elongated philtrum, thin upper lip and protruding ears were noticeable [Figure - 1].
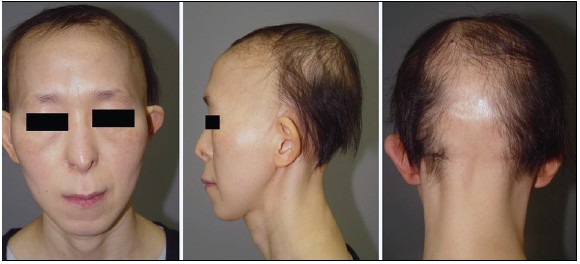 |
| Figure 1: The typical trichologic and craniofacial features of TRPS I and receded medio-occipital hairline of roughly triangular shape |
Clinical examination revealed clinodactyly and brachydactyly with racket thumbnails, Perthes-like change of the hips [Figure - 2] and sparse axillary and pubic hair. Several years ago, she underwent reconstructive orthopedic and plastic surgeries of phalanges of both hands and nose. Radiologic examination showed hypoplastic left femur and hip joint compared to the right side with slight anterior bowing of proximal shaft of femur [Figure - 3], as well as premature osteoarthritis with sclerosis in bilateral sacro-iliac joints. Cone-shaped epiphyses of the phalanges suggesting longstanding inflammatory polyarthritis with secondary osteoarthritic change were noted.
 |
| Figure 2: Lateral deviation, clinodactyly and brachydactyly with racket thumb nails and splitting, Perthes-like change of the hips despite several reconstructive orthopedic and plastic surgeries |
 |
| Figure 3: Corresponding radiologic findings of skeletal abnormalities of TRPS I |
Laboratory tests, including complete blood cell count, liver function test, thyroid function test, blood coagulation test, antinuclear antibody and urinalysis were within normal limits or negative.
Histopathologic examination of the scalp was performed by incisional skin biopsy and revealed atrophic pilosebaceous unit and significantly reduced hair follicles without inflammation or scarring [Figure - 4]. On light and polarizing microscopy (Hitachi FE-SEM S-4700® ), the hair shafts embedded in mounting media showed neither reduced diameter nor structural defects [Figure - 5].
 |
| Figure 4: Atrophic pilosebaceous unit and significantly reduced hair follicles without inflammation or scarring (H and E, ×40, ×100) |
 |
| Figure 5: No abnormal structural findings in scanning electron microscopic examination |
Mutation analysis was performed by direct sequencing of genomic DNA isolated from peripheral blood leukocytes and identified a transition of cytosine to thymine at position 1630 (exon 4), which results in amino acid change R544X and a premature stop of translation [Figure - 6]. This nonsense mutation was previously reported and listed on the website of the Human Gene Mutation Database (HGMD® ).
 |
| Figure 6: A transition of cytosine to thymine at position 1630 (exon 4), which results in amino acid change R544X and a premature stop of translation |
The family gene study was unavailable because they did not consent to test for the mutation analysis, but no suspicious sign was detected in her three generation members so our patient seemed to be a sporadic case.
After diagnosis as a case of TRPS I, we decided to observe her without any specific treatment and recommended that she receive regular orthopedic and endocrinologic evaluation.
Dicussion
TRPS I is a rare autosomal dominant hereditary syndrome characterized by trichologic, craniofacial and skeletal abnormalities with high penetrance and variable expressivity. [6]
Developmental defects of the hair include fine, sparse and slow-growing hair, reaching only a limited length. [7] Sparse hair ranges from almost normal hair to severe diffuse alopecia; in very rare cases, completely baldness. [8] The receded fronto-temporal hairline and laterally scanty eyebrow are usually observed, and, less often, the eyelash may also be involved. [7],[8] The receded medio-occipital hairline of roughly triangular shape was observed in only one familial case. [6] Our case is assumed to be a sporadic case with receded medio-occipital hairline of roughly triangular shape. Sparseness of hairs may also affect the axillary and pubic hair. [7]
Craniofacial abnormalities include a bulbous pear-shaped nose, elongated philtrum, thin upper lip and maxillary prognathism with mandibular hypoplasia. [9] These craniofacial changes are variably expressed so sometimes these are very subtle, especially after reconstructive plastic surgery as in our case. Careful evaluation of suspicious cases to identify potential affected carriers may be needed.
Abnormalities of selected bones include a cone-shaped epiphyses of the middle phalanges, which are the most frequent finding, brachydactyly and brachytarsia, deviation of the proximal and (less frequently) distal phalanges, painless swelling of the proximal interphalangeal joints and hip changes. [6] Almost all cases with TRPS I were reported with short stature of variable expressivity. Less frequently, dental anomalies, spinal scoliosis, lordosis and kyphosis are observed. These skeletal abnormalities, especially hip changes, are not infrequent, and are risk that factors affect the outcome, not merely a cosmetic problem.
Compatible with the clinical features, the TRPS I gene is highly expressed within the affected organs of the patients, including cartilage, developing joints, hair follicles and the nasal region. [10] A recent study revealed that mice with a disrupted TRPS I gene develop a chondrodysplasia characterized by impaired chondrocyte proliferation and decreased apoptosis of growth plate chondrocytes caused by increased expression and activation of Stat3. [11] The underlying pathologic mechanisms are not fully understood.
The scanning electron microscopy (SEM) findings are divided between alterations of the cuticular pattern and hair shaft structure and no abnormalities in TRPS I. [6],[12],[13] Our patient also showed neither reduced diameter nor structural defects.
Several reports described an association of TRPS I with endocrine disturbances, including diabetes mellitus, hypothyroidism, and growth hormone deficiency. [14]
TRPS1 gene was first identified in 2000 and mapped on chromosome 8q23.39. It includes 7 exons and encodes a polypeptide of 1,281 amino acids (3843 bp.) with two potential nuclear localization signals and an unusual combination of nine putative zinc-finger motifs, including IKAROS-like and GATA-binding sequences. [2],[3] The IKAROS-like sequence contains the last two motifs and mediate the transcriptional repressive function of the TRPS I gene. [15] Two nuclear localization signals flank the GATA zinc-finger region, one of which was related to prevent the translocation of mutant TRPS1 to the nucleus. [15] To date, more than 50 mutations including deletion, nonsense and missense mutations in the gene have been found.
We assumed that, because of the great variable expressivity of clinical features, many cases of TRPS I remain underdiagnosed until more severely, and typically affected family members consult a physician for medical care. [6] The sparse hair is often the most disturbing sign for which the patients seek medical advice. [12] So it is necessary to consider TRPS I when dermatologists see a patient who complains of sparse hair with subtle structural abnormalities.
Even if there is no established treatment, through the careful evaluation of suspicious cases to identify potential mutation carriers, the patient can receive information about the disease and genetic counseling.
| 1. |
Giedion A. Das tricho-rhino-phalangeale Syndrom. Helv Paediatr Acta 1966;5:475-82.
[Google Scholar]
|
| 2. |
Momeni P, Glöckner G, Schmidt O, von Holtum D, Albrecht B, Gillessen-Kaesbach G, et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat Genet 2000;24:71-4.
[Google Scholar]
|
| 3. |
Piccione M, Niceta M, Antona V, Di Fiore A, Cariola F, Gentile M, et al. Identification of two new mutations in TRPS 1 gene leading to the tricho-rhino-phalangeal syndrome type I and III. Am J Med Genet A 2009;149:1837-41.
[Google Scholar]
|
| 4. |
Lüdecke HJ, Wagner MJ, Nardmann J, La Pillo B, Parrish JE, Willems PJ, et al. Molecular dissection ofa contiguous gene syndrome: Localization of the genes involved in the Langer-Giedion syndrome. Hum Mol Genet 1995;4:31-6.
[Google Scholar]
|
| 5. |
Lüdecke HJ, Schaper J, Meinecke P, Momeni P, Gross S, von Holtum D, et al. Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet 2001;68:81-91.
[Google Scholar]
|
| 6. |
Seitz CS, Lüdecke HJ, Wagner N, Bröcker EB, Hamm H. Trichorhinophalangeal syndrome type I: Clinical and molecular characterization of 3 members of a family and 1 sporadic case. Arch Dermatol 2001;137:1437-42.
[Google Scholar]
|
| 7. |
Camacho F, Armijo M, Naranjo R, Dulanto F. Le syndrome Tricho-Rhino-Phalangien (Giedion). Ann Dermatol Venereol 1978;105:17-21.
[Google Scholar]
|
| 8. |
Besser F, Wells RS. The tricho-rhino-phalangeal syndrome (a family with three affected members). Br J Dermatol 1976;95:39-41.
[Google Scholar]
|
| 9. |
George S, Pulimood S, Korah I. Trichorhinophalangeal syndrome type I. J Eur Acad Dermatol Venereol 1998;11:66-8.
[Google Scholar]
|
| 10. |
Chen LH, Ning CC, Chao SC. Mutations in TRPS1 gene in trichorhinophalangeal syndrome type I in Asian patients. Br J Dermatol 2010;163:416-9.
[Google Scholar]
|
| 11. |
Suemoto H, Muragaki Y, Nishioka K, Sato M, Ooshima A, Itoh S, et al. Trps1 regulates proliferation and apoptosis of chondrocytes through Stat3 signaling. Dev Biol 2007;312:572-81.
[Google Scholar]
|
| 12. |
Prens EP, Peereboom-Wynia JD, de Bruyn WC, van Joost T, Stolz E. Clinical and scanning electron microscopic findings in a solitary case of trichorhinophalangeal syndrome type I. Acta Derm Venereol 1984;64:249-53.
[Google Scholar]
|
| 13. |
Van Neste D, Dumortier M. Tricho-rhino-phalangeal syndrome. Disturbed geometric relationships between hair matrix and dermal papilla in scalp hair bulbs. Dermatologica 1982;165:16-23.
[Google Scholar]
|
| 14. |
Sohn YB, Ki CS, Park SW, Cho SY, Ko AR, Kwon MJ, et al. Clinical, biochemical, and genetic analysis of two korean patients with trichorhinophalangeal syndrome type I and growth hormone deficiency. Ann Clin Lab Sci 2012;42:307-12.
[Google Scholar]
|
| 15. |
Kaiser FJ, Brega P, Raff ML, Byers PH, Gallati S, Kay TT, et al. Novel missense mutations in the TRPS1 transcription factor define the nuclear localization signal. Eur J Hum Genet 2004;12:121-6.
[Google Scholar]
|
Fulltext Views
3,248
PDF downloads
1,123





![[Figure - 1]](#fig_ijdvl_2014_80_1_54_125515_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2014_80_1_54_125515_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2014_80_1_54_125515_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2014_80_1_54_125515_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2014_80_1_54_125515_f5.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2014_80_1_54_125515_f6.jpg){kind=link}