Translate this page into:
Clinicopathological correlation of acquired hypopigmentary disorders
2 Delhi Dermatology Group, Delhi Dermpath Laboratory, New Delhi, India
Correspondence Address:
Asha Kubba
10, Aradhana Enclave, R.K. Puram, Sector 13, New Delhi - 110 066
India
| How to cite this article: Patel AB, Kubba R, Kubba A. Clinicopathological correlation of acquired hypopigmentary disorders. Indian J Dermatol Venereol Leprol 2013;79:376-382 |
Abstract
Acquired hypopigmentary disorders comprise a significant group of disorders that affect Indians and Asians. The pigment disturbance in darker skin individuals can be very distressing to the patient and the family. These disorders cover a wide array of pathologies including infections, autoimmune processes, lymphoproliferative disorders, and sclerosing diseases. Histological diagnosis is particularly important because treatments for these diseases are varied and specific. This review will focus on histopathological diagnosis based on clinicopathological correlation for commonly encountered disorders such as leprosy, vitiligo, lichen sclerosus, pityriasis alba (PA), and pityriasis versicolor (PV). Atypical or uncommon clinical presentation of classic diseases such as hypopigmented mycosis fungoides (HMF) and hypopigmented sarcoidosis are also included.Introduction
Acquired hypopigmentary disorders comprise a significant group of disorders that affect Indians and Asians. The pigment disturbance in darker skin individuals can be very distressing to the patient and the family. These disorders cover a wide array of pathologies including infections, autoimmune processes, lymphoproliferative disorders, and sclerosing diseases. Histological diagnosis is particularly important because treatments for these diseases are varied and specific.
This review will focus on histopathological diagnosis based on clinicopathological correlation for commonly encountered disorders such as leprosy, vitiligo, lichen sclerosus, pityriasis alba (PA), pityriasis versicolor (PV). Atypical or uncommon clinical presentation of classic diseases such as hypopigmented mycosis fungoides (HMF) and hypopigmented sarcoidosis are included.
Vitiligo
Vitiligo is characterized by spontaneous appearance of sharply demarcated, depigmented patches that accentuate under Wood′s light. Clinical subtypes include focal, acrofacial, segmental and the most common, nonsegmental form, also known as vitiligo vulgaris [1] [Figure - 1]a. Vitiligo has known autoimmune associations more so in the nonsegmental form . [2]
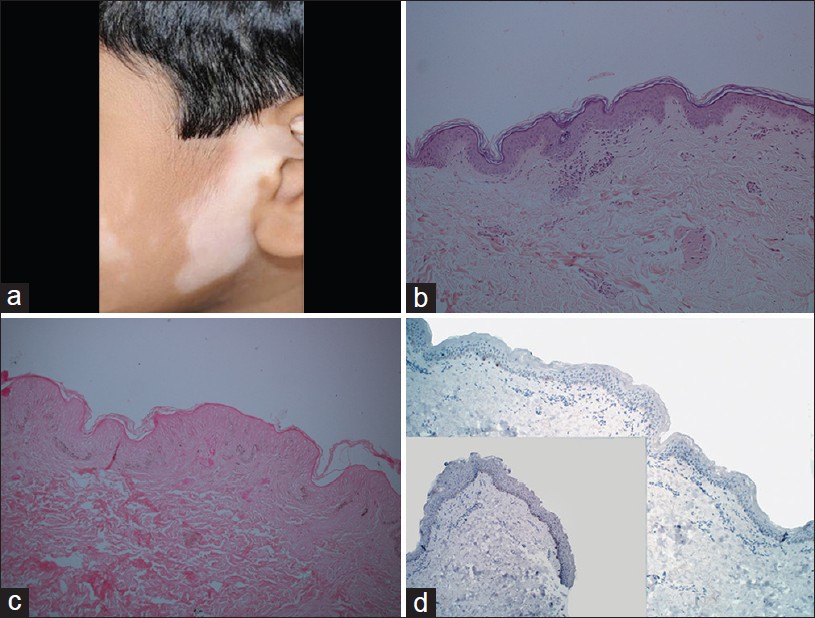 |
| Figure 1: Vitiligo: (a) A hypopigmented patch on the face. (b) Lymphohistiocytic inflammation at interface (H and E, ×100). (c) Hypomelanotic epidermis (Masson Fontana, ×100). (d) Absence of melanocytes in vitiligo lesion with control skin as inset (Melan-A stain, ×100) |
Histopathological features of vitiligo vary according to the life of the lesion. The best practice for diagnostic evaluation of a pigmentary disorder is to compare lesional skin with perilesional normal skin of the patient.
An early, evolving lesion of vitiligo shows subtle pathological features that provide a diagnostic challenge. Hematoxylin- and eosin-stained (H and E) section shows basal hypomelanosis, confirmed by Masson Fontana stain. Sparse to mild lymphocytic inflammation in papillary dermis in contrast to perilesional skin. Sometimes, lichenoid interface dermatitis may be present. [3] Prominent lymphocytic infiltrate was observed in a single case report of vitiligo mimicking HMF . [4] Additional features like acanthosis, exocytosis, spongiosis, and melanophages are not diagnostic. Demonstration of reduced number of melanocytes by Melan-A, a melanocyte-specific immunohistochemical marker, is diagnostic [Figure - 1]b-d.
An established lesion of vitiligo shows scant inflammatory infiltrate, complete loss of melanocytes along with complete loss of epidermal pigmentation. Light microscopy findings are supported by Dopa and silver nitrate negative staining, and by monoclonal and polyclonal antibodies directed against melanocytes. [5] Degenerative changes in keratinocyte and melanocyte have been documented. [3],[5],[6]
In a review of 74 vitiligo specimens, [3] histopathological findings observed were: (a) absence of pigment and suprabasal vacuolization in all cases; (b) inflammatory changes were seen in lesions of shorter duration; (c) degenerative changes in long-standing cases; (d) suprabasal clear cells in perilesional skin in 50% of specimens; (e) perivascular inflammatory infiltrate in 30%; (f) thinned epidermis in 53%; (g) sweat gland degeneration in 72%, and (h) dermal nerve degeneration in 78% cases. Based on this study, loss of pigment and dermal inflammation are important diagnostic features to look for.
The differential diagnosis of vitiligo includes nevus depigmentosus (ND) in which loss of melanin and reduction of melanocytes are much less as compared to vitiligo. [7] PA also shows hypomelanosis but reduction in the melanocyte number is variable. Lichen sclerosus shows lichenoid features with papillary dermal sclerosis.
Hansen′s Disease
The incidence of Hansen′s disease is declining worldwide but it is still frequently encountered in India, Eastern Africa, and Brazil. From our experience, it is often biopsied as the clinical and social implications are considered grave. Hypopigmented, anesthetic patch (s) with feathered borders, and thickened regional nerve are virtually diagnostic of the disease. Erythematous, indurated plaques with or without anesthesia generate a wider differential diagnosis and demand pathological evaluation. Indeterminate lesions also show variable anesthesia and can be challenging to diagnose clinically. [8] Hansen′s disease and its reactions constitute a wide morphological spectrum earning for the disease the sobriquet "the great mimic."
Histology varies depending on the host immune response to the organism. This is the basis for the classification scheme of Hansen′s disease. Tuberculoid leprosy shows granulomatous inflammation comprised of epithelioid histiocytes and Langhan′s giant cells, surrounded by lymphocytes in periadnexal and perineural locations. The infiltrate can appear linear because of its perineural distribution and organisms are extremely hard to identify. In borderline variants, there is a grenz zone sparing the papillary dermis, as opposed to the papillary dermal involvement in the tuberculoid type. The tuberculoid form also has better delineated granulomatous nodules, increased proportion of lymphocytes, and fewer organisms. Indeterminate leprosy is difficult to diagnose both clinically and histologically as it shows a perivascular lymphohistocytic infiltrate with periadnexal and perineural foci. Organisms are rarely visualized . Lesion of nonspecific dermatitis may be distinguished from early leprosy by the presence of epidermal hyperplasia, numerous plasma cells, and sparing of cutaneous appendages/dermal nerves. As an endemic disease, leprosy is included in the clinical differential diagnosis of hypomelanotic lesions. In the absence of acid fast bacillus, indeterminate type is difficult to prove histologically. Clinical anesthesia is an important clue in favor of leprosy.
Lichen Sclerosus et Atrophicus
The preferred term is "Lichen sclerosus" as hypertrophic lesions also occur. [9] Lichen sclerosus presents as hypopigmented, atrophic papules, and plaques, [Figure - 2]a that progressively develop to wrinkled, parchment-like depressed scar. In the early stages, the lesions may be lichenoid in character and may show a shiny scale. Single or multiple lesions may arise anywhere on the torso and limbs. Anogenital lichen sclerosus et atrophicus (LSA) is a common and distinctive subset affecting middle-aged individuals, women more often than men. LSA at times is indistinguishable from vitiligo and mandates histological evaluation. [10]
 |
| Figure 2: Lichen sclerosus: (a) Irregular white plaque, trunk. (b) Squamatized basal layer, papillary dermal sclerosis, lichenoid infiltrate (H and E, ×100) |
Histology of a fully developed lesion shows hyperkeratosis, plugged ostia, thin effaced epidermis with vacuolar damage, and a squamatized epidermis. Additional features are thickened basement membrane, edema, hyalinization of the papillary dermis, and telangiectasia. A lymphohistiocytic inflammatory infiltrate is pushed below the hyalinized area. Subepidermal edema is sometimes prominent [Figure - 2]b. A lichen planus-like interface pathology may be seen in early lesions or at the edge of fully evolved lesion. [11] Basilar epidermotropism and thickened basement membrane help distinguish LSA from lichen planus. [12] Absence of interface damage and presence of hypertrophic collagen fibers in deep dermis suggest morphea. Papillary dermal inflammation and edema/sclerosis are not observed in vitiligo lesion.
Hypopigmented Mycosis Fungoides
HMF affects dark skin individuals mostly. Single or multiple scaly hypopigmented patches develop insidiously on the trunk and extremities. Children and young adults comprise a majority in the reported cases [Figure - 3]a. Clinically, HMF mimics vitiligo, PA, leprosy, and pityriasis versicolor. [13] There are several case reports of vitiligo-like HMF. [14],[15] In a review of clinicopathological characteristics of patients with HMF from Mumbai, the mean time to diagnosis was over 3.5 years, underscoring the need for early biopsy in this clinical setting. [16]
 |
| Figure 3: Hypopigmented Mycosis fungoides: (a) Persistent patches of hypopigmentation on trunk. (b) Epidermotropic lymphocytes in non-spongiotic epidermis, papillary dermal fibrosis infiltrated by lymphocytes (H and E, ×100). (c) Epidermotropic CD8 positive T-cell phenotype (Immunohistochemical stain, ×100) |
HMF histology is similar to that of classic mycosis fungoides (MF). Diagnosis of patch stage MF is based on architectural features and not so much on cellular atypia. Marked epidermotropism in a non-spongiotic epidermis is the clue. Spongiosis, however, is often seen but with disproportionate number of lymphocytes in the epidermis in a psoriaisform or lichenoid pattern. The key diagnostic features are haloed lymphocytes, tagging of lymphocytes at the dermo-epidermal junction, associated papillary dermis fibrosis, and wiry bundles of collagen that are infiltrated by lymphoid cells including some atypical cells [Figure - 3]b. Pautrier microabscesses are seldom seen in early lesions, and are, therefore, not essential for the diagnosis. Melanin is reduced in the basal epidermis and melanophages may be seen in papillary dermis. On immunohistochemical analysis, the epidermotropic lymphocytes, in a HMF lesion, are predominantly of CD8 positive phenotype [17] [Figure - 3]c. Further, these epidermotropic CD8 positive lymphocytes were found to have altered T-cell receptor gamma chain gene for rearrangement consistent with malignant T-cell clone . [17] Classic MF is, predominantly, CD4 positive lymphoproliferative disease. In pediatric age group, CD8 positive T-cell phenotype was present in majority of cases. [17] Diagnosis of patch stage MF cannot be made without a biopsy and often requires more than one biopsy. Selecting a poikilodermatous lesion for biopsy is much more rewarding than a scaly patch lesion. Epidermotropism is not a feature of PA, pityriasis versicolor or leprosy. Pityriasis lichenoides lesions among children can sometimes display epidermotropism, which then requires immunohistochemical markers to exclude HMF.
Hypopigmented Sarcoidosis
This variant is reported mainly in black South Africans. [18] Skin lesions are variable and range from shiny, irregular, hypochromic, macules, papules, and nodules distributed extensively on limbs and trunk [Figure - 4]a and b. Ichthyosiform and ulcerative forms [18] and hypochromic macules localized over deep dermal and subcutaneous granulomas have also been reported. [19] Electron microscopy suggests toxic damage to melanocytes [19],[20] which may be reversed by PUVA therapy. [21]
 |
| Figure 4: Hypochromic sarcoidosis: (a) Shiny, irregular hypochromic papules and macules, upper back neck. (b) Similar patches on forearms, bilateral |
Histopathology is similar to classic sarcoidosis. Non-caseating epithelioid granulomas in the dermis and in periadnexal location are observed. Discrete, rounded, typically naked granulomas, may show scattering of lymphocytes, are often surrounded by condensed collagen fibers. Variable number of Langhan′s type giant cells are present. Fibrinoid necrosis was found in 12% of biopsies in a series of 54 cases. [18] Caseation necrosis is rare. Inclusion bodies such as Shaumann body and Asteroid body may be seen but are not pathognomonic. These are found in other granulomatous disorders including tuberculosis and berylliosis. [22] Sarcoidal granulomas must be distinguished from infective granulomas of tuberculoid leprosy, cutaneous tuberculosis, and non-infective granulomas. Polaroscopic examination for foreign material is a good practice. Special stains for acid fast bacillus and fungal stains are required. Identification of mycobacterial DNA in 16 of 20 cases of cutaneous sarcoidosis has pathogenetic implications. [23] Histopathological diagnosis remains one of exclusion.
Post-kala-azar Dermal Leishmaniasis
Post-kala-azar dermal leishmaniasis (PKDL) is a late sequele of visceral leishmaniasis caused by the organism Leishmania donovani. It occurs 1-3 years after the systemic illness and has been reported mostly from the Indian subcontinent and Sudan. PKDL is thought to be a cutaneous immunological reaction in the dermis to an attenuated parasite turned dermotropic from being viscerotropic, following treatment. Skin lesions are distributed over face, trunk, and limbs. Polymorphic morphology includes hypopigmented macules, papules, and nodules. Erythema and induration along the butterfly area of face may accompany the classic lesions. [24],[25]
Histology of a nodular lesion shows a dense infiltrate of lymphocytes, histiocytes, and plasma cells in the upper dermis with an occasional eosinophil. Follicular plugging is prominent in facial lesions. Intracellular LD bodies are typically sparse and require diligent search. The organisms are better visualized with Weigert iron hematoxylin or Giemsa stains. Macular lesions, on the other hand, show subtle histopathology with minimal to mild upper dermal inflammatory infiltrate of lymphohistiocytes, plasma cells. LD bodies are rare. These features are not diagnostic of PKDL. Most sensitive and specific test is polymerase chain reaction (PCR). [26] Fite stain is a must to exclude leprosy. Histopathological features and PCR test would distinguish PKDL from vitiligo.
Pityriasis Versicolor
PV is commonly seen in tropical climates and is caused by over-colonization of the stratum corneum with Malassezia furfur, a lipophilic yeast. In Caucasian skin, these lesions are fawn-colored, but in colored skin, the lesions are commonly hypopigmented. [13] The hypopigmented lesions in caucasians are known as pityriasis versicolor alba (PVa). [27] Clinically, PV presents as oval to circular macules, showing feathered borders and fine scaling, distributed in seborrhoeic areas, and appearing discrete or coalescing to form large, irregular sheets. Under Wood′s light, PV may show equivocal accentuation. An unusual variant of hypopigmented confluent reticulate papillomatosis seen in dark skin individuals clinically mimics PV. [28]
Histopathology shows spores and hyphae in upper layers of stratum corneum on H and E stain but best observed with PAS stain. [29] Mild superficial, perivascular lymphocytic infiltrate is noted. There is no pigment incontinence. Clinical basis of hypomelanosis is a result of low-density melanin in the epidermis with normal number of melanocytes, highlighted by Masson Fontana stain. Electron microscopy shows morphologically altered melanocytes with cytoplasmic and mitochondrial vacuoles, and reduced number of melanosomes in the keratinocytes. [29] Lichen sclerosus has distinctive histology. Vitiligo shows loss of melanocytes.
Idiopathic Guttate Hypomelanosis
It is a hypopigmentary disorder of unknown cause that presents with sharply circumscribed, white, 2-6 mm macules, on the shins and occasionally on extensor aspects of forearms [Figure - 5]a. Idiopathic guttate hypomelanosis (IGH) lesions are persistent and asymptomatic. They may be mistaken for vitiligo and LSA.
 |
| Figure 5: Idiopathic guttate hypomelanosis: (a) Discrete hypopigmented atrophic macules on the limb. (b) Hypomelanotic epidermis, no inflammation, (H and E, ×100). (c) Sharp demarcation between hypomelanotic and normally pigmented epidermis (Masson Fontana stain, ×100) |
Histopathology shows epidermal atrophy with flattening of rete ridges, covered by orthokeratosis. Hypomelanosis of IGH is best visualized with Masson Fontana stain [Figure - 5]b and c. The epidermal melanin content of the lesional area is sharply reduced compared to adjacent normally pigmented epidermis. Dopa stain shows reduction or partial loss of melanocytes. [30] Differential diagnosis includes vitiligo, leprosy, and lichen sclerosus. Most of these can be diagnosed on H and E. Melan-A, a specific immunohistochemical marker, can demonstrate complete loss of melanocytes in vitiligo.
Pityriasis Alba
PA, a common, benign condition, may be clinically confused with HMF and Hansen′s disease, especially when it occurs on extra facial sites. In the study from South India, PA was the most common hypopigmentary disorder noted in the pediatric population. [13] It typically shows ill-defined hypopigmented patches with minimal fine scale on the face or trunk. [31] PA is associated with atopic dermatitis.
Histology of an early lesion of P. alba shows follicular dilatation, plugging, spongiosis, superficial perivascular infiltrate, and dermal edema. Late lesion shows nonspecific changes of hyperkeratosis, focal parakeratosis, and spongiosis. There is a variable reduction of melanin and melanocytes in the basal epidermis. Ultrastructurally, degenerative changes in melanocytes and reduced number of melanosomes within keratinocytes were seen. [32] Histopathological evaluation may be required to distinguish it from PV and leprosy.
Lichen Planus Depigmentosus
This is a term coined by the authors to refer to a subset of vitiligo patients who evoke lichenoid characteristics, namely, involvement of volar wrists, dorsa of feet, hands, and trunk [Figure - 6]a subtle lichenoid morphology and interface pathology in the initial phase. Histologically, observed lichenoid tissue reaction was centered at the dermo-epidermal junction, and included basal cell damage and a lichenoid infiltrate of lymphohistiocytes (personal observation) [Figure - 6]b. In time, the aforementioned features disappear and the condition progressively becomes indistinguishable from vitiligo vulgaris. The authors believe that this hitherto unreported entity is a distinct subset of lichen planus that koebnerizes into vitiligo and represents the opposite counterpart of lichen planus pigmentosus.
 |
| Figure 6: Lichen planus depigmentosus: (a) Irregular depigmented macules with lichenoid sheen, shoulder. (b) Lichenoid features at the interface with hypomelanosis (H and E, ×100) |
Nevus Depigmentosus
ND presents at birth or later in childhood. Clinically, single to multiple well-demarcated hypopigmented macules are commonly seen on back and buttocks. Other affected sites are chest, abdomen, face, neck, and arms [Figure - 7]a. Thus, ND can be mistaken for childhood vitiligo and segmental vitiligo.
 |
| Figure 7: Nevus depigmentosus: (a) Well demarcated hypopigmented macular patch on the flank. (b) Non-inflammatory, no dermal pigment (H and E, ×100). (c) Epidermal hypomelanosis (Masson Fontana stain, ×100) |
Histopathology of ND is nonspecific on H and E. Dermal inflammation and melanophages are uncommon. Decrease in melanin compared to nonlesional skin is highlighted by Masson Fontana stain [Figure - 7]b and c. Reports vary on whether or not the number of melanocytes is decreased in lesional skin, but a study of 30 cases of ND showed that the melanocyte count was overall stable with only a decrease in melanin. [33] Basal hypomelanosis and mild dermal inflammation may provide clue to diagnosis of vitiligo. Melan-A immunohistochemical stain would be confirmatory.
Conclusion
The above account of acquired hypopigmentary disorders briefly covers most of the common entities. It underscores the importance of pathological evaluation in clinching diagnoses such as hypopigmented cutaneous T-cell lymphoma (CTCL) and hypopigmented sarcoidoses. It highlights the part played by pathology in evaluating and managing interface dermatoses in which pigmentary disturbances are consequence or sequel. Pathology is also useful in eliminating entities that may be confused with common hypopigmentary disorders such as vitiligo, ND, and IGH.
| 1. |
Handa S, Kaur I. Vitiligo: Clinical findings in 1436 patients. J Dermatol 1999;26:653-7.
[Google Scholar]
|
| 2. |
Pradhan V, Patwardhan M, Thakkar V, Kharkar V, Khopkar U, Ghosh K, et al. Vitiligo patients from India (Mumbai) show differences in clinical, demographic and autoantibody profiles compared to patients in western countries. J Eur Acad Dermatol Venereol 2011. [In press]
[Google Scholar]
|
| 3. |
Alikhan A, Felsten LM, Daly M, Petronic-Rosic V. Vitiligo: A comprehensive overview Part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J Am Acad Dermatol 2011;65:473-91.
[Google Scholar]
|
| 4. |
Horn TD, Abanmi A. Analysis of the lymphocytic infiltrate in a case of vitiligo. Am J Dermatopathol 1997;19:400-2.
[Google Scholar]
|
| 5. |
Kovacs SO. Vitiligo. J Am Acad Dermatol 1998;38:647-66.
[Google Scholar]
|
| 6. |
Bhawan J, Bhutani LK. Keratinocyte damage in vitiligo. J Cutan Pathol 1983;10:207-12.
[Google Scholar]
|
| 7. |
Kim YC, Kim YJ, Kang HY, Sohn S, Lee ES. Histopathologic features in vitiligo. Am J Dermatopathol 2008;30:112-6.
[Google Scholar]
|
| 8. |
Piris A, Lobo AZ, Moschella SL. Global dermatopathology: Hansen�s disease-current concepts and challenges. J Cutan Pathol 2010;37:125-36.
[Google Scholar]
|
| 9. |
Neill SM, Lewis FM, Tatnall FM, Cox NH, British association of dermatologists. British association of dermatologists� guidelines for the management of lichen sclerosus 2010. Br J Dermatol 2010;163:672-82.
[Google Scholar]
|
| 10. |
Attili VR, Attili SK. Vitiligoid lichen sclerosus: A reappraisal. Indian J Dermatol Venereol Leprol 2008;74:118-21.
[Google Scholar]
|
| 11. |
Calonje E, Neill S, Bunker C, Francis N, Chaux A, Cubilla A. Lichen sclerosus. In: Calonje E, Brenn T, Lazar A, McKee PH, editors. McKee's Pathology of the Skin. 4 th ed. Philadelphia: Elsevier Saunders; 2012. p. 452-6.
th ed. Philadelphia: Elsevier Saunders; 2012. p. 452-6.'>[Google Scholar]
|
| 12. |
Weedon D. Lichen sclerosus et atrophicus. In: Weedon D, editor. Weedon's Skin Pathology. 3 rd ed. London: Churchill Livingstone; 2010. p. 313-6.
rd ed. London: Churchill Livingstone; 2010. p. 313-6.'>[Google Scholar]
|
| 13. |
Sori T, Nath AK, Thappa DM, Jaisankar TJ. Hypopigmentary disorders in children in South India. Indian J Dermatol 2011;56:546-9.
[Google Scholar]
|
| 14. |
Ngo JT, Trotter MJ, Haber RM. Juvenile-onset hypopigmented mycosis fungoides mimicking vitiligo. J Cutan Med Surg 2009;13:230-3.
[Google Scholar]
|
| 15. |
Das JK, Gangopadhyay AK. Mycosis fungoides with unusual vitiligo-like presentation. Indian J Dermatol Venereol Leprol 2004;70:304-6.
[Google Scholar]
|
| 16. |
Khopkar U, Doshi BR, Dongre AM, Gujral S. A study of clinicopathologic profile of 15 cases of hypopigmented mycosis fungoides. Indian J Dermatol Venereol Leprol 2011;77:167-73.
[Google Scholar]
|
| 17. |
El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, McCalmont TH. Hypopigmented mycosis fungoides: Frequent expression of a CD8+T-cell phenotype. Am J Surg Pathol 2002;26:450-7.
[Google Scholar]
|
| 18. |
Jacyk WK. Cutaneous sarcoidosis in black South Africans. Int J Dermatol 1999;38:841-5.
[Google Scholar]
|
| 19. |
Hubler WR Jr. Hypomelanotic canopy of sarcoidosis. Cutis 1977;19:86-8.
[Google Scholar]
|
| 20. |
Clayton R, Breathnach A, Martin B, Feiwel M. Hypopigmented sarcoidosis in the negro. Report of eight cases with ultrastructural observations. Br J Dermatol 1977;96:119-25.
[Google Scholar]
|
| 21. |
Patterson JW, Fitzwater JE. Treatment of hypopigmented sarcoidosis with 8-methoxypsoralen and long wave ultraviolet light. Int J Dermatol 1982;21:476-80.
[Google Scholar]
|
| 22. |
Calonje E. Sarcoidosis. In: Calonje E, Brenn T, Lazar A, McKee PH, editors. McKee's Pathology of the Skin. 4 th ed. Philadelphia: Elsevier Saunders; 2012. p. 281-8.
th ed. Philadelphia: Elsevier Saunders; 2012. p. 281-8.'>[Google Scholar]
|
| 23. |
Li N, Bajoghli A, Kubba A, Bhawan J. Identification of mycobacterial DNA in cutaneous lesions of sarcoidosis. J Cutan Pathol 1999;26:271-8.
[Google Scholar]
|
| 24. |
Ganguly S, Das NK, Barbhuiya JN, Chatterjee M. Post-kala-azar dermal leishmaniasis: An overview. Int J Dermatol 2010;49:921-31.
[Google Scholar]
|
| 25. |
Singh N, Ramesh V, Arora VK, Bhatia A, Kubba A, Ramam M. Nodular post-kala-azar dermal leishmaniasis: A distinct histopathological entity. J Cutan Pathol 1998;25:95-9.
[Google Scholar]
|
| 26. |
Ramesh V, Ramam M, Singh R, Salotra P. Hypopigmented post-kala-azar dermal leishmaniasis. Int J Dermatol 2008;47:414-6.
[Google Scholar]
|
| 27. |
Thoma W, Krämer HJ, Mayser P. Pityriasis versicolor alba. J Eur Acad Dermatol Venereol 2005;19:147-52.
[Google Scholar]
|
| 28. |
Hudacek KD, Haque MS, Hochberg AL, Cusack CA, Chung CL. An unusual variant of confluent and reticulated papillomatosis masquerading as tinea versicolor. Arch Dermatol 2012;148:505-8.
[Google Scholar]
|
| 29. |
Galadari I, el Komy M, Mousa A, Hashimoto K, Mehregan AH. Tinea versicolor: Histologic and ultrastructural investigation of pigmentary changes. Int J Dermatol 1992;31:253-6.
[Google Scholar]
|
| 30. |
Falabella R. Idiopathic guttate hypomelanosis. Dermatol Clin 1988;6:241-7.
[Google Scholar]
|
| 31. |
Vinod S, Singh G, Dash K, Grover S. Clinico epidemiological study of pityriasis alba. Indian J Dermatol Venereol Leprol 2002;68:338-40.
[Google Scholar]
|
| 32. |
In SI, Yi SW, Kang HY, Lee ES, Sohn S, Kim YC. Clinical and histopathological characteristics of pityriasis alba. Clin Exp Dermatol 2009;34:5917.
[Google Scholar]
|
| 33. |
Lee HS, Chun YS, Hann SK. Nevus depigmentosus: Clinical features and histopathologic characteristics in 67 patients. J Am Acad Dermatol 1999;40:21-6.
[Google Scholar]
|
Fulltext Views
5,629
PDF downloads
1,357





![[Figure - 1]](#fig_ijdvl_2013_79_3_376_110800_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2013_79_3_376_110800_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2013_79_3_376_110800_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2013_79_3_376_110800_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2013_79_3_376_110800_f5.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2013_79_3_376_110800_f6.jpg){kind=link}
![[Figure - 7]](#fig_ijdvl_2013_79_3_376_110800_f7.jpg){kind=link}