Translate this page into:
Epidermolysis bullosa pruriginosa: A rare presentation with asymptomatic lesions
2 Department of Medicine, PGIMS, Rohtak, Haryana, India
Correspondence Address:
Sangita Ghosh
42\136, New Ballygunge Road, Kolkata - 39
India
| How to cite this article: Ghosh S, Chaudhuri S, Jain VK. Epidermolysis bullosa pruriginosa: A rare presentation with asymptomatic lesions. Indian J Dermatol Venereol Leprol 2013;79:235-237 |
Abstract
Epidermolysis bullosa pruriginosa (EBP) is a subtype of dominant dystrophic epidermolysis bullosa (DDEB) and is clinically characterized by pruritic lichenified plaques or prurigo-like lesions with violaceous linear scarring. Pruritus has always been described as one of the most striking features in EBP. Mutations in COL7A gene, especially in the glycine residue, have been shown to cause this form of DDEB. In this report, we describe a north Indian familial clustering of three cases of EBP, spread across two generations, presenting with hypertrophic lichenoid cutaneous lesions, which were completely asymptomatic. Clinical and histopathological analysis favored the diagnosis of EBP in all three cases. They are being reported for their unusual asymptomatic presentation.Introduction
Epidermolysis Bullosa Pruriginosa (EBP) is a rare, inherited, distinctive clinical subtype of Dystrophic Epidermolysis Bullosa (DEB), caused by mutations in the COL7A1 gene which encodes type VII collagen, the major structural component of anchoring fibrils at the dermal-epidermal junction. [1] Ultrastructurally, there is sublamina densa level of blister formation. [2] This is a rare disease with fewer than 100 patients reported in the literature till date. Various inheritance patterns have been described, including autosomal dominant, autosomal recessive and sporadic patterns. It is characterized clinically by severe pruritus, lichenified plaques or prurigo like lesions, violaceous linear scarring, occasional trauma-induced blistering, excoriations, milia, nail dystrophy and in some cases, albopapuloid lesions on trunk. [2] This form of inherited epidermolysis bullosa may not clinically develop until adult life, leading to confusion regarding it′s inherited nature. [3]
Case Reports
Case 1
The index case is a 35-year-old male, who presented with asymptomatic hypertrophic prurigo-like papules on both lower extremities for the past 20 years, associated with progressive dystrophy of all the nails. Patient gave history of intermittent trauma-induced acral blistering in childhood, which used to heal with papules and scarring. Patient had never experienced any pruritus or any other symptoms over these lesions. He was the eldest of six siblings and four among them, as well as his parents, were asymptomatic; only his younger brother had similar lesions. On examination, there were multiple lichenified prurigo-like papules, nodules and plaques, associated with scarring and occasional milia, on ventral aspects of bilateral lower legs extending up to the knees. All nails were dystrophied with irregular thickening and pigmentation of nail plates and shedding of nails, leaving only vestigial nail spurs.
Case 2
A 3-year-old boy, 3 rd son of index case, presented with history of trauma-induced blistering on dorsa of both knees, since 1 year of age. For the past 3 months, active blistering has stopped and he now has two asymptomatic post-inflammatory, well defined hypopigmented healed plaques, one on each knee, with hypertrophic scarring on left knee plaque. Nails of both big toes were dystrophied. Rest of the nails, mucosa, hair, teeth, palms and soles were normal. His other two siblings (sister aged 7 years and brother aged 5 years) are asymptomatic till date [Figure - 1] a-d.
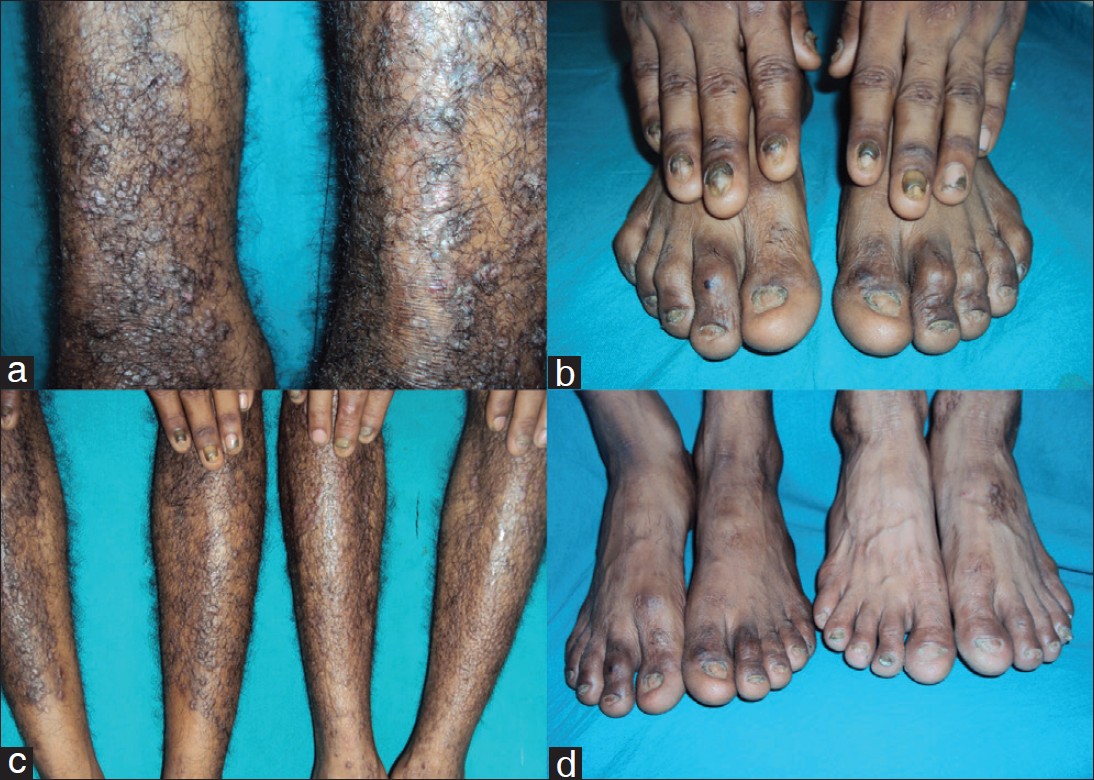 |
| Figure 1: (a) Lichenoid, hypertrophic, prurigo like papules and plaques on bilateral legs of case 1 (b) Bilateral finger and toe nails dystrophy of index case. (c) Both case 1 and case 3 having Epidermolysis Bullosa Pruriginosa on both legs and bilateral fingernails dystrophies. (d) Both case 1 & case 3, with bilateral finger and toe nails dystrophies |
Case 3
A 28-year-old male, brother of index case, gave history of similar non-pruritic, hypertophic prurigo-like papules and nodules in both lower legs, associated with dystrophy of multiple finger nails and toe nails without any mucosal involvement. He too, like his elder brother (case 1) has never had pruritus and experienced mechanical trauma induced acral blistering in his childhood. His two sons (aged 6 and 8 years) are asymptomatic till date [Figure - 2]a and b.
 |
| Figure 2: (a) Case 2 with well-defined post-inflammatory hypopigmented plaques over bilateral knees with hypertrophic scarring on left knee. (b) Case 2 with bilateral toe nails dystrophy |
All three patients were born out of non-consanguineous marriages and had no lesions at birth. Mode of inheritance pointed towards an autosomal dominant pattern [Figure - 3]. Neither of them had history of atopy nor any underlying systemic or cutaneous diseases. Mucosa, teeth, palms and soles, in all the three cases, were uninvolved. None of them had albopapuloid lesions. Systemic examination findings and baseline biochemical parameters were within normal limits. Histopathological examination of lesional skin from each patient had a similar picture with demonstrable hyperkeratosis, acanthosis with subepidermal bullae along with inflammatory infiltrates in the dermis [Figure - 4]a and b. Direct immunofluorescence testing of lesional skin was negative for Immunoglobulin G (IgG), Immunoglobulin M (IgM), or C 3 in all of them. Electron microscopy and gene mutation studies could not be done due to limited resources. They were started on topical tacrolimus, but the patients were lost to follow-up after 3 weeks of therapy.
 |
| Figure 3: Pedigree chart showing affected index case (case 1), his brother (case 2) and their four other unaffected siblings, all born out of non-consanguineous marriage of unaffected parents. Case 3 is the third child of index case and his siblings were unaffected |
 |
| Figure 4: (a) (H and E, ×5) Hyperkeratosis, acanthosis with a prominent sub-epidermal separation with inflammatory infiltrates and a milium in the dermis (b) (H and E, ×10) Hyperkeratosis, acanthosis with sub-epidermal separation |
Discussion
EBP is a subtype of dystrophic epidermolysis bullosa, characterized by a combination of severe pruritus and skin fragility that lead to scarring as well as hypertrophic, lichenified, prurigo like nodules and plaques. [3] Typically lesions mostly occur on the shins, and also on other parts of the legs, forearms, elbows, dorsal aspect of hands, shoulders, and lower back. [2] The face and flexures are always spared, and nail dystrophy, albopapuloid lesions, blisters and milia are other common but variable features. [4] Age of onset of skin lesions in EBP is very variable; development of the first clinical sign in adulthood is not uncommon although the reason for the delayed presentation is not known. Lesions may develop at birth. [5] to as late as 40 years of age. [3] Differential diagnoses that need to be ruled out before one can conclusively narrow down to this diagnosis are lichen simplex chronicus, hypertrophic lichen planus, lichen amyloidosis, prurigo nodularis, dermatitis artifacta among others. History of mechanical blistering, family history of similar lesions, characteristic morphology of cutaneous lesions with nail dystrophy and finally histopathological demonstration of sub epidermal separation, are arguments in favor of the diagnosis of EBP. Ultrastructurally, there is a sublamina densa level of blister formation due to defect in the anchoring fibrils at the dermal-epidermal junction. [2] Study of the molecular basis of dominant dystrophic EB (classical) and EBP shows that both diseases are caused by a missense glycine mutation, substitution mutation by different amino acids in the same codon of COL 7A (G2028R and G2028A). [6] Predicting the clinical course is very difficult because, apart from the genetic mutation, additional environmental, metabolic, immunological, hormonal and other cutaneous or systemic factors lead to the initiation of EBP. Phenotypic appearances may vary between affected individuals or families, but pruritus has been a uniform features in all and it appears that the patient either experiences an increased level of pruritus or an increased excoriation response to normal pruritus, perhaps due to development of immediate type hypersensitivity to environmental allergens. [7] These cases are of considerable clinical interest because of their lack of pruritus, which may deter clinical suspicion and prompt diagnosis. Clinical management of EBP is often difficult and treatment is usually unsatisfactory and includes general measures for pruritus like anti-histaminics and topical steroid with or without occlusion. [8] Trials of a few interventions which have been found to have a variable response in EBP are topical tacrolimus, [4] systemic ciclosporin, [9] Dapsone, [10] thalidomide, [7] and cryotherapy. [5]
| 1. |
Fine JD, Eady RA, Bauer EA, Briggaman RA, Bruckner-Tuderman L, Christiano A, et al. Revised classification system for inherited epidermolysis bullosa: Report of the Second International Consensus Meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol 2000;42:1051-66.
[Google Scholar]
|
| 2. |
McGrath JA, Schofield OM, Eady RA. Epidermolysis bullosa pruriginosa: Dystrophic epidermolysis bullosa with distinctive clinicopathological features. Br J Dermatol 1994;130:617-25.
[Google Scholar]
|
| 3. |
Ee HL, Liu L, Goh CL, McGrath JA. Clinical and molecular dilemmas in the diagnosis of familial epidermolysis bullosa pruriginosa. J Am Acad Dermatol 2007;56:S77-81.
[Google Scholar]
|
| 4. |
Banky JP, Sheridan AT, Storer EL, Marshman G. Successful treatment of epidermolysis bullosa pruriginosa with topical tacrolimus. Arch Dermatol 2004;140:794-6.
[Google Scholar]
|
| 5. |
Das JK, Sengupta S, Gangopadhyay AK. Epidermolysis bullosa pruriginosa - Report of three cases. Indian J Dermatol Venereol Leprol 2005;71:109-11.
[Google Scholar]
|
| 6. |
Murata T, Masunaga T, Shimizu H, Takizawa Y, Ishiko A, Hatta N, et al. Glycine substitution mutations by different amino acids in the same codon of COL7A1 lead to heterogeneous clinical phenotypes of dominant dystrophic epidermolysis bullosa. Arch Dermatol Res 2000;292:477-81.
[Google Scholar]
|
| 7. |
Ozanic Bulic S, Fassihi H, Mellerio JE, McGrath JA, Atherton DJ. Thalidomide in the management of epidermolysis bullosa pruriginosa. Br J Dermatol 2005;152:1332-4.
[Google Scholar]
|
| 8. |
Yesudia PD, Krishnan S, Jayaraman M, Janaki VR, Yesudian P. Epidermolysis bullosa pruriginosa. Indian J Dermatol Venereol Leprol 2000;66:249-50.
[Google Scholar]
|
| 9. |
Yamasaki H, Tada J, Yoshioka T, Arata J. Epidermolysis bullosa pruriginosa (McGrath) successfully controlled by oral cyclosporin. Br J Dermatol 1997;137:308-10.
[Google Scholar]
|
| 10. |
Qayoom S, Masood Q, Sultan J, Hassan I, Jehangir M, Bhat YJ, et al. Epidermolysis bullosa: A series of 12 patients in kashmir valley. Indian J Dermatol 2010;55:229-32.
[Google Scholar]
|
Fulltext Views
2,646
PDF downloads
339





![[Figure - 1]](#fig_ijdvl_2013_79_2_235_107645_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2013_79_2_235_107645_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2013_79_2_235_107645_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2013_79_2_235_107645_f4.jpg){kind=link}