Translate this page into:
Reticulate pigmentary disorders
Correspondence Address:
Kabir Sardana
466 sector 28, NOIDA, UP
India
| How to cite this article: Sardana K, Goel K, Chugh S. Reticulate pigmentary disorders. Indian J Dermatol Venereol Leprol 2013;79:17-29 |
Abstract
Reticulate pigmentary disorders is a term that is loosely defined to include a spectrum of acquired and congenital conditions with different morphologies. The presentations vary from the reticular or net like pattern to the" freckle like" hyper and hypopigmented macules that are usually restricted to the true genetic "reticulate" pigmentary disorders. There is little clarity on this topic and related terms, in major dermatology textbooks. Hence, to harmonize the different entities we feel that the term "mottled pigmentation" could be used to include reticulate pigmentary disorders (acquired and congenital), dyschromasias and the disorders with a reticular pattern. The genetic reticulate pigmentary disorders can also be classified according to the gene loci which in the majority of cases are localized to keratin 5/14. A more useful clinical method of classification is based on the regional distribution, which includes facial, truncal, acral or flexural types. In this review we will largely focus on the inherited reticulate pigmentary disorders.Introduction
The topic of reticulate pigmentation and its related terminology [Box 1] has been covered in major textbooks [1],[2],[3],[4],[5],[6],[7],[8] but an overlapping and indiscriminate use of the terms [Box 1] leads to a lack of clarity on this important entity. Reticulate pigmentary disorders are a group of disorders which includes the inherited reticulate pigmentary disorders, [6],[7] which present with hyperpigmented macules with a morphology reminiscent of "freckles" with varying pigment and size. Disorders with acquired "reticulate" or "reticular/net like′′ pattern of pigmentation have a morphology that is unlike the classic "freckle like" hyperpigmentation seen in inherited reticulate pigmentary disorders. Dyschromatoses [1],[3],[5],[6],[7],[8] is another term that encom-passes conditions with both hyperpigmented and hypopigmented macules, many of which are small in size and irregular in shape. A third entity that often presents with reticulate or more accurately reticular pigmentation are the poikilodermatous conditions [6],[7] wherein the telangiectasias are less pronounced than the fine net like pigmentation. To encompass all these entities mottled pigmentation. [1],[2],[3],[4],[5],[6],[7],[8] could be an appropriately christened term which by definition includes all disorders with a variable hue, size and shape of pigmentation

Classification
Till date there is no clear consensus [1],[2],[3],[4],[5],[6],[7],[8] regarding the spectrum and definition of the term "reticulate pigmentation" even in speciality textbooks. [7] The vari-ous term used for these disorders are given in Box 1 and have been used interchangeably which makes the nosology difficult to comprehend. The true reticulate disorders classically include dyskeratosis congenita, Dowling Degos, acropigmentation of Kitamura, Naegeli Franceschetti Jadassohn (NFJ) syndrome, X linked reticulate pigmentary disorder, dyschromatosis sym-metrica hereditaria and dyschromatosis universalis hereditaria. [8] The acquired disorders [Figure - 1], [Table - 1] have a pattern of reticulate pigmentation with macules of a size larger than the "freckle" like morphology of the true genetic reticulate pigmentary disorders. A minority of the acquired disorders have the classic "net like"/"reticular" pattern and include lichen planus pigmentosus, Riehl′s melanosis [Figure - 2], erythema ab igne [Figure - 3], cutis marmorata, livedo reticularis and post inflammatory hyperpigmentation [2] amongst other conditions [Table - 1]. Some of the acquired conditions [Table - 1] have an admixture of hyperpigmented and hypopigmented macules which is strictly not in consonance with the definition of reticulate pigmentation [Box 1]. To encompass the differing morphology of the acquired and genetic forms of reticulate pigmentation [1],[2],[3],[4],[5],[6],[7],[8] the umbrella term, mottled pigmentation [Box 1] is ideal and includes in addition to the true/genetic reticulate disorders, acquired reticular/reticulate conditions, poikilodermas and dyschromatoses [Figure - 1] and [Table - 1].
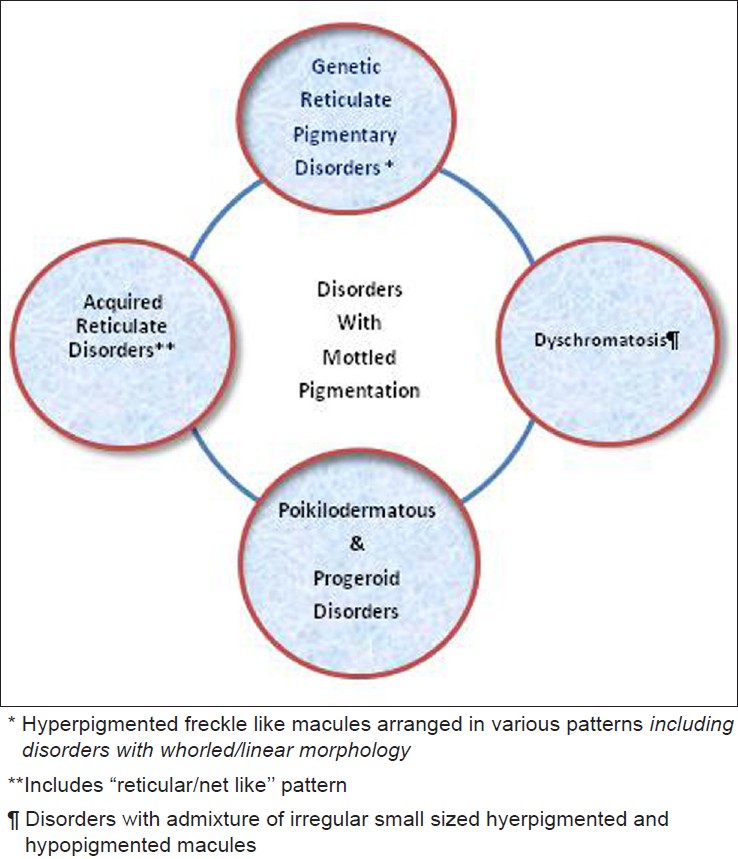 |
| Figure 1: Spectrum of "Mottled" Pigmentary Disorders |
 |
| Figure 2: Reticulate pigmented macules on the face in a case of pigmented contact dermatitis (Riehl's melanosis) |
 |
| Figure 3: A case of Erythema ab igne with reticulate hyperpigmentation due to exposure to heat |

Though most of the true reticulate dermatoses are thought to be inherited, the genes responsible for their expression have not been definitively identified [2] Wherever genetic analysis has been done it has thrown up some interesting loci which can help "lump" entities [9],[10] into one spectrum instead of splitting them into various types [Figure - 4] a and b. Most of the commonly seen genetic reticulate pigmentary disorders have defects localized to keratin 5 and keratin 14 gene. It has now been confirmed that NFJ/DPR [Figure - 4]a are consequent to mutation of the KRT14 gene which results in haploinsufficiency and predisposes the keratinocyte to proapoptic stimuli. [5],[6] Similar defects in keratin 5 predisposes to epithelial remodeling, melanosome mistargetting and plays an important role in melanosome transport. [5],[6] These points to a defect in the keratinocyte melanocyte synergy which may consequentially affect the pigment regulation of melanocytes. [5],[6] The similar morphology of Dowling Degos disease, acropigmentation of Kitamura and EBS with mottled pigmentation is also explained by their common gene loci [Figure - 4]a. Conversely, the conventional "lumping" of DUH (dyschromatosis universalis hereditaria) and DSH (dyschromatosis symmetrica hereditaria) is not backed by genetic loci [Figure - 4]a though they are morphologically similar.
 |
| Figure 4: (a) A depiction of gene and chromosome loci in selected disorders with reticulate pigmentation[1],[2],[3],[4],[5],[6],[7],[8] (http://www.ncbi.nlm.nih. gov/omim). (b) A depiction of gene and chromosome loci on X chromosome in selected disorders with reticulate pigmentation[1],[2],[3],[4],[5],[6],[7],[8],[12] (http://www.ncbi.nlm.nih.gov/omim) |
Clinically, the reticulate pigmentary disorders can be classified based on the extent and distribution as localized, generalized, flexural or acral [Figure - 5].This distribution pattern can serve as a useful way to diagnose the conditions which have a predilection for certain body areas. A subset [Figure - 5] of reticulate pigmentary disorders (dyskeratosis congenita, Mendes de Costa syndrome and dermatopathia pigmentsosa reticularis/NFJ syndrome) are associated with features of ectodermal dysplasia, the presence of which provides a clue to their diagnosis .The salient features of the main disorders are summarized in [Table - 2] and [Table - 3] [1],[2],[3],[4],[5],[6],[7],[8],[11],[12],[13],[14] which also serves as a differential diagnosis of the reticulate pigmentary disorders.
 |
| Figure 5: A depiction of the regional distribution of reticulate pigmentary disorders |


Our primary focus will be on the disorders with a genetic defect which largely fit into the classic morphology of true reticulate pigmentation and dyschromatosis.
Genetic Reticulate Pigmentary Disorders
Reticulate acropigmentation of Kitamura
This autosomal dominant condition has been largely reported from the Asian countries. [1],[3],[6],[7] There are a few reports of familial cases [15] and the genetic loci [Figure - 4] a overlaps with Dowling Degos disease
Clinical features
This gradually progressive disorder has a onset in the first to second decade of life. [16] The lesions are slightly depressed, sharply demarcated black/brown macules localized to the dorsum of the hands and feet [Figure - 6]a.These increase in number and spread centripetally with age. The presence of small pits that cause a break in the epidermal ridge patterns [Figure - 6]b on the palms and rarely on the dorsum of fingers, is a diagnostic feature. [17] Eventually the extensor aspects of the limbs, neck, upper trunk, face [Figure - 7] and eyelids are affected. It can rarely involve the flexures and the palms and soles. Uncommonly disseminated hypo- or depigmented macules and papules have also been reported. [18]
 |
| Figure 6: (a) and (b) Reticulate acropigmentation of Kitamura with depressed hyperpigmented macules on dorsa of bilateral hands and palmar pits with breaks in the epidermal ridge pattern |
 |
| Figure 7: "Freckle like" hyperpigmented macules involving the face and the neck, in a case of reticulate acropigmentation of Kitamura |
Histologically, hyperpigmented lesions show epider-mal atrophy, elongation and increased melanin in of rete ridges, and increased numbers of DOPA-positive melanocytes. [2],[19]
Treatment and prognosis
There is very little that can change the course of the pigmentation. Treatment with 20% azelaic acid resulted in remarkable reduction of hyperpigmentation in one patient. [20]
Acropigmentation symmetrica of Dohi (dyschromatosis symmetrica hereditaria, DSH)
This autosomal dominant disorder [21] commonly described in Asians has an onset during childhood. There is a pathological mutation of the double-stranded RNA-specific adenosine deaminase gene (ADAR1 or DSRAD) and the location is at the 1q21.1-q21.2 [22] [Figure - 4]a. This enzyme plays an important role in post-transcriptional modification of the messenger RNA (RNA editing). [22] Impaired RNA editing affects the differentiation of melanoblasts during melanogenesis into hyper and hypoactive melanocytes. [22] Consequentially, the affected melanoblasts are those that emigrate the farthest, namely the hands and feet, thus accounting for the distribution of the disorder. [22]
Clinical features
The lesions begin in the first to second decade and are classically non progressive .The skin findings are characterized by hypo- and hyper-pigmented macules on the dorsal and ventral aspects of the hands [Figure - 8] and feet, which may extend to the proximal portions of the limbs (knees and elbows). Similar lesions (freckle-like macules) can be found on the face. [3],[5],[6],[7],[13]
 |
| Figure 8: Freckle like hypo and hyperpigmented macules on the palms in a case of dyschromatosis symmetrica hereditaria (DSH) |
The histology of the lesions shows either increased or decreased basilar pigmentation in the hyperpigmented or hypopigmented lesions respectively. Occasional pigment incontinence is seen in hypopigmented lesions. [2]
Treatment and prognosis
Surgical therapy with transplantation of thin split-thickness skin autografts has been tried. [23] Q-switched ruby, Q-switched Nd:YAG, Q-switched alexandrite, and the 510 mm pulsed dye lasers have been used to treat hyperpigmented macules especially on face. [13]
Dyschromatosis universalis hereditaria (DUH)
Dyschromatosis universalis symmetrica hereditaria was originally believed to have a localized acral form, dyschromatosis symmetrica hereditaria (reticulated acropigmentation of Dohi). [13],[14] A recent study though has clarified that genetically it is different from dyschromatosis symmetrica hereditaria (DSH) with the defect localized to 12q21-q23 loci [22],[24] [Figure - 4]a. It has an autosomal dominant pattern of inheritance and has been reported from Japan and India. [5],[7]
Clinical features
It has onset in early childhood and is characterized by mottled pigmentation which originates from the hands and can progress to involve the trunk, extremities and the face. The lesions are characterized by hyperpigmented macules admixed with hypopigmented lesions [Figure - 9] and can also involve the palms, soles and oral mucosa. [1],[3],[5],[6],[7],[13],[14] The nail are hyperpigmented dystrophic with pterygium formation being the classic finding. Various associations that have been reported [7],[25] include coxa valga, nerve compression, small stature, high-tone deafness, photosensitivity and neurosensory hearing defect.
 |
| Figure 9: Truncal involvement with hypo and hyperpigmented macules in a case of dyschromatosis universalis hereditaria (DUH) |
Treatment and prognosis
As the condition is non-progressive, counseling seems to be the best available option. Targeting the pigmented lesion with the Q-switched alexandrite laser is an option [26] but recurrence is inevitable.
Unilateral dermatomal pigmentary dermatosis (UDPD)
It is a segmental form of dyschromatosis which has been included under the same spectrum as DUH and DSH. In UDPD, the distribution is segmental. It is differentiated from segmental neurofibromatosis and partial unilateral lentiginosis by the mottled hyperpigmentation and hypopigmentation. [27]
Dermatopathia pigmentosa reticularis
It is an autosomal dominant disorder that classically has a generalized distribution. The diagnostic triad is of reticulate hyperpigmentation, noncicatricial alopecia of the scalp, eyebrows, axillae, and onychodystrophy. [1],[3],[5],[7],[14],[28] Genetic analysis has revealed a close association with NFJ syndrome and EBS with mottled pigmentation [28] [Figure - 4]a.
Clinical features
The hyperpigmentation is generalized, and is most prominent on the trunk and proximal extremities. The presence of small "confetti" like macules gives rise to a characteristic reticulate pattern. The nails show onychodystrophy and loss of nails, with formation of pterygium [Figure - 10]a during the second year of life. There is progressive alopecia involving the scalp, eyelashes, eyebrows and axilla. Other features include hypohidrosis or hyperhidrosis, punctuate palmoplantar keratoses, absent dermatoglyphics, and non-scarring acral blisters. [14] Fine punctate spots on the cornea and brown pigmentation of the bulbar conjunctiva have been also reported [Figure - 10]b.
 |
| Figure 10: (a) A patient of dermatopathia pigmentosa reticularis with reticulate pigmentation over dorsum of both hands and associated dystrophy of finger nails with thin lusterless nails. (b) Also seen are fine punctuate corneal opacities and brown pigmentation of bulbar conjunctiva |
A very close differential is NFJ syndrome [28],[14] [Table - 2]. DPR has marked alopecia, absence of dermatoglyphics, hyperhidrosis and pigmentation of the mucosa with onychodystrophy. NFJ on the contrary manifests with dental defects with little or no nail dystrophy. Also there is a tendency of fading of pigment by adolescence. Recent genetic analysis reveals that they are probably manifestations of the same genotypical defect [Figure - 4]a.
Histology reveals clumps of melanin-laden melanophages seen in the papillary dermis in a patchy distribution without overlying epidermal hyperpigmentation. [2]
Treatment and prognosis
There is no known treatment for the pigmentary disorder. [7] The associated keratoderma though has been successfully treated with etretinate. [29]
Dowling-Degos disease and its variants
Syn: Dark dot disease, reticular pigment anomaly of flexures
This is a progressive, disorder of pigmentation, charac-terized by flexural, pigmented reticulate macules and comedone-like papules on the back and neck. [1],[3],[5],[7],[14] It is inherited as an autosomal dominant trait [1],[2] and the gene defect is believed to be localized to kertain. [5],[30] It is usually sporadic though some affected families have been reported.
As the localization of the gene [Figure - 4]a overlaps with a related condition it is also referred to as Dowling Degos - Kitamura disease. [7],[10]
Clinical features
The onset is delayed with an onset in early adulthood (30-40 years). The macules gradually become confluent in a "lacelike" or reticulate pattern. Reticulate pigmentation of the flexures is seen which is gradually progressive and symmetrical. The initial sites affected are the groins and the axillae [Figure - 11]a and the pigmentation insidiously progresses to involve the neck, inframammary creases, trunk, proximal arms and the antecubital fossae. Acneiform perioral pits [Figure - 11]b and comedones are the hallmark of the disease. There are also comedolike, hyperkeratotic follicular papules on the neck and axillae and epidermal cysts. [1],[3],[5],[7],[14] The hyperkeratotic papules are thought to arise in areas of friction. Other associated findings include hidradenitis suppurativa, [31] keratoacanthoma [32] and squamous cell carcinoma. [31] Hypopigmented macules on the trunk can be seen as a association in some cases. [18]
 |
| Figure 11: (a) A case of Dowling Degos with keratotic papules and hyperpigmented macules on the axilla. (b) Note the presence of perioral acneiform pits |
Histologically, the characteristic finding is of an atrophic epidermis with long, narrow, branched rete ridges that intertwine at their bases ("antler-like" appearance). [33] Increased melanin in the basal membrane and dilated follicles with cysts can also be seen.
Treatment and prognosis
There is no effective therapy. Many topical medications including azelaic acid, retinoic acid, hydroquinone and corticosteroids, as well as systemic retinoids, have been tried with minimal improvement. [1],[3],[6],[7] However, there is a single report of the use of the Er:YAG laser with pulse energy of 1,000 and 1,200 mJ, which after three consecutive passes led to favorable results. [34] Probably the atrophic lesions could respond with the use of fractional lasers.
Variants of Dowling Degos disease [Figure - 5]
Haber′s syndrome is a disorder characterized by pigmented keratotic papules on the axilla, neck, and torso with pitted scars on the face and persistent facial erythema. [35] Galli-Galli disease is essentially an acantholytic variant of DDD. [36] Pigmentatio reticularis faciei et colli is possibly a variant of DDD which presents with hyperpigmentation of the face and neck with multiple epidermoid cysts. [37]
Dyskeratosis congenita
Syn: Zinsser-Cole-Engman syndrome
This disorder is commonly a X-linked recessive disorder [38] though AD [39] and AR [40] inheritance have also been reported [Figure - 4]b The defective genes include dyskerin, TERC, TERT, NHP2, and NOP10. The genetic defect in the X-linked form is located on Xq28 (DKC1 gene for dyskerin). [41] Autosomal-dominant inheritance is often associated with mutations in htr (hterc). [39]
Clinical features
There is a lacy reticulate telangiectatic hyper-pigmentation interspersed with areas of hypopig-mentation [Figure - 12]a seen on the face, neck, trunk and upper thighs. [38] Atrophy and cyanosis of the dorsal aspects of the hands and feet with hyperkeratosis and hyperhidrosis of palms and soles may also be present. The mucosa shows leukokeratosis which may involve the pharynx, anorectal and urogenital mucosae. The nails are dystrophic with pterygium formation [Figure - 12]b and the hair is thin, lusterless and sparse.
 |
| Figure 12: (a) Case of dyskeratosis congenita with lacy reticulate pigmentation of neck with associated telangiectasias. (b) Also seen is onychodystrophy of finger nails and pterygium formation |
The extracutaneous manifestations include early dental loss or extensive caries, aplastic anemia with bleeding problems and purpura, esophageal diverticuli with dysphagia, retardation of growth, hypogonadism and mental retardation.
Hoyeraal-Hreidarsson syndrome is probably a variant characterized by intrauterine growth retardation, cerebellar hypoplasia, mental retardation, microcephaly, progressive combined immune deficiency, and aplastic anemia. [42] The syndrome is genetically heterogeneous though some patients demonstrate DKC1 gene mutations and are therefore allelic to dyskeratosis congenita.
Another severe variant of DKC is Revesz syndrome [7],[8],[38],[39] characterized by exudative retinopathy, leucoplakia, nail pits, sparse hair, CNS defects and aplastic anemia. The gene defect lies in chromosome 14q12 and TINF2 gene.
Treatment and prognosis
The disorder has a a poor prognosis and death is the rule, often in the third decade. Associated malignancies include Hodgkin′s disease, oropharyngeal, esophageal, gastric, and pancreatic carcinomas, and squamous cell carcinomas. The cause of mortality includes malignant neoplasms, infection by opportunistic agents, failure of bone marrow and hematological malignancies (mainly in the second or third decade).
Fanconi anemia
Syn: Familial pancytopenia or familial panmyelophthisis
This is a autosomal-recessive condition with five complementation groups (FA-A, FA-B, FA-C, FA-D, and FA-E) [43] which affect hematopoiesis. FA-A has been localized to 16q24.3, and FA-D to 3p22.26.
Clinical features
The skin has diffused mottled pigmentation (hypo-pigmentation, hyperpigmentation, and café-au- lait macules). [44] The other findings include absence of the thumbs, aplasia of the radius, severe hypoplastic anemia, thrombocytopenia, retinal hemorrhage, strabismus, generalized hyperreflexia and testicular hypoplasia.
The syndrome is associated with increased risk of myelomonocytic leukemia, squamous cell carcinoma, and hepatic tumors. Human papillomavirus DNA is often found in the squamous cell carcinomas. Both cutaneous and pulmonary manifestations of associated Sweet′s syndrome have been reported.
Miscellaneous Genetic Reticulate Pigmentary Disorders
These are some of the rare conditions encountered in clinical practice and are incorporated for the sake of completion of text. [5],[7],[8],[13],[14]
Acromelanosis albo-punctata (ectodermal dysplasia)
This condition is characterized by diffuse hyper-pigmentation with superimposed atrophic guttate hypomelanosis seen primarily on the dorsal aspects of the hands and flexures. Other features include keratotic follicular papules on the legs, pili torti, platynychia.
Berlin syndrome (ectodermal dysplasia)
This has been described in one family of Iranians living in Israel. The patients had generalised melanoleucoderma with ectodermal dysplasia (hypodontia, hypotrichosis), short stature, furrowing around the mouth and eyes, pokiloderma around the joints, sexual underdevelopment in male patients and mental retardation.
Lucky/Winter syndrome (ectodermal dysplasia)
This was described in two unrelated children with features of ectodermal dysplasia, generalized hyperpigmentation with superimposed guttate hypo-melanosis on the flexures. Other features seen were scant lightly pigmented hair, enamel hypoplasia (single central incisor in one patient), digitalized thumb and short stature.
Ziprowski-Margolis syndrome
This X linked disorder [Figure - 4]b is also known as the albinism and deafness syndrome. In this there is pigmentary dilution of the hair and skin except for the buttock and genitalia. With time pigmented macules are seen over this giving the "leopard like" appearance. Other features include deaf mutism and heterochromia irides.
Wende-Bauckus/Pegum syndrome
The disease has an onset at one year of age and presents with a background hyperpigmentation (gray-brown) which is more pronounced on the trunk than on the extremities. The superimposed white macules which are confluent on the flexures give it the "mottled" appearance.
Chédiak-Higashi syndrome (CHS) and Griscelli syndrome (GS)
Though these are characterized by pigmentary dilution of the skin, they may also have diffuse hyperpigmentation with superimposed guttate hypopigmentation in sun-exposed areas. Other findings include bleeding diathesis (CHS), neurologic abnormalities (CHS, GS1), immunodeficiency (CHS, GS2).
Acquired Reticulate Pigmentary Disorders
A detailed list is given in [Table - 1], but we will focus on two conditions which present primarily with a macular morphology and thus mimic the morphology of genetic reticulate disorders.
Confluent and reticulate papillomatosis (CRP)
This eponymous entity is named after two French dermatologists, Gougerot and Carteaud, [45] and it has a variable clinical presentation. This condition is commoner in females than males.
Clinical features
The onset of the disease is around 20 years of age. The lesions are red, verrucous, minimally scaly papules, occuring in the inframammary, interscapular and epigastric regions, which coalesce to form brown plaques. [5],[7],[13],[14],[45] There is accentuation in the neck and in the axillae. With progression, the lesions acquire the characteristic reticulate appearance.
The criteria for diagnosis are. [46]
- Hyperpigmented papules and plaques involving the chest and/or back with a reticulated peripheral margin.
- No hyphae suggestive of tinea versicolor present on potassium hydroxide preparation or skin biopsy.
- Histological changes including hyperkeratosis, papillomatosis, acanthosis alternating with mild atrophy, and evidence of mild dermal inflammation and vasodilatation.
Treatment and prognosis
Topical agents include tacalcitol and calcipotriol. The treatment of choice for CRP is minocycline, [47] which is given in a dose of 100-200 mg per day for weeks to months. Other antibiotics tried include clarithromycin (500 mg daily for five weeks), erythromycin (1000 mg daily) and azithromycin (500mg daily). Isotretinoin and etretinate are also effective drugs. [48]
Prurigo pigmentosa
It is a rare inflammatory skin disease of unknown pathogenesis and is characterized by pruritic, erythematous urticarial papules, papulovesicles, or vesicles that are symmetrically localized on the trunk and nape and that resolve quickly leaving behind reticulate pigmentation. [7],[49] Lesions in different stages are usually present together and disease has a waxing waning course. [49] It is seen most commonly in young Japanese women. Various causative factors have been implicated such as ethnic predisposition, environmental causes, seasonal variation, mechanical stimuli, contact allergens, drugs such as bismuth subsalicylate containing antacid, Helicobacter pylori infection, atopic disease etc. [50],[51] A comprehensive clinico-histopathological diagnostic criteria have been given by Boer et al. [49]
Histopathological features range from superficial peri-vascular dermatitis, spongiotic dermatitis, and lichenoid dermatitis to post-inflammatory hyperpigmentation, depending on the stages of skin lesions. [49]
Various medications have been tried with variable results. Antihistamines and steroids have been found to be ineffective whereas minocycline, tetracycline and doxycycline have shown promising results. [48] Other established treatments include sulfonamides (dapsone, sulfamethoxazole), macrolide antibiotics (clarithromycin, roxythromycin), potassium iodide, and isotretinoin. [52]
Conclusion
A practical clinical classification based on regional distribution can help the clinician to diagnose most of the genetic reticulate disorders [Figure - 5] but probably genetic analysis might be a more rational way of analyzing these conditions [Figure - 4]a and b. This [Figure - 4]a and b has shown that probably the reticulate disorders have variable phenotypical expressions of similar gene defects.The therapy of most of the genetic disorders is inadequate and it is practically impossible to ameliorate the defect. Presently, the clinician can at best counsel the patients about the invariably benign course of the disorder.
The acquired causes are simpler to diagnose [Table - 1] and may not pose a problem as they are usually associated with the primary dermatoses in most cases. The acquired conditions are equally frustrating to treat but as they have a more dominant primary cutaneous morphology which is of primary concern, the secondary reticulate pigmentation is less of an issue.
We believe that a rational classification of these disorders [Box 1] necessitates using the overarching term "mottled pigmentation" which would include the "true/genetic" reticulate pigmentary disorders [Figure - 6], [Figure - 7], [Figure - 8], [Figure - 9], [Figure - 10], [Figure - 11] [Figure - 12] which are different morphologically from the acquired disorders [Figure - 2] and [Figure - 3].
| 1. |
James WD, Berger TG, Elston DM. Disturbances of Pigmentation; Pigmented Anomalies of the Extremities. Andrews' Diseases of the Skin Clinical Dermatology. 11 th ed. China: Elsevier's; 2006. p. 855-6.
th ed. China: Elsevier's; 2006. p. 855-6. '>[Google Scholar]
|
| 2. |
Kovarik CL, Spiehogel RL, Kantor GR. Pigmentary Disorders of Skin. In: Lever WF, Elder, Elder DE, editors. Lever's Text Book of Histopathology. 2 nd ed. New Delhi: Wolters Kluwer; 2009. p. 689-90.
nd ed. New Delhi: Wolters Kluwer; 2009. p. 689-90. '>[Google Scholar]
|
| 3. |
AV Anstey. Disorders of Skin Color. In: Burns DA, Breathnach SM, Cox N, Griffith CS, editors. Rook's Textbook of Dermatology. 8 th ed. Oxford: Wiley Blackwell; 2010. p. 58.20-2.
th ed. Oxford: Wiley Blackwell; 2010. p. 58.20-2. '>[Google Scholar]
|
| 4. |
Lapeere H, Boone B, Schepper SD. Hypomelanoses and hypermelanoses. In: Wolff K, Goldsmith LA, Katz S, Gilchrist BA, Paller A, Leffell DJ, editors. Fitzpatrick Dermatology in General Medicine. 7 th ed. New York: McGraw Hill; 2008. p. 631-40.
[Google Scholar]
|
| 5. |
Bolognia JL. Disorders of hypopigmentation and hyperpigmentation. In: Harper J, Orange H, Prose N, editors. Textbook of Pediatric Dermatology. 2 nd ed. United States: Blackwell Science; 2000. p. 868-70.
[Google Scholar]
|
| 6. |
Ramrath K, Stolz W. Disorders of Melanin Pigmentation. In: Burgdorf, Plewig, Wolff, Landthaler, editors. Braun Falco's Dermatology. 3 rd ed. Newyork: Springer; 2009. p. 772-4.
rd ed. Newyork: Springer; 2009. p. 772-4.'>[Google Scholar]
|
| 7. |
Esterly NB, Basalge E. Genetic Epidermal Syndromes: Disorders characterized by Reticulated Hyperpigmentation. In: Nordlund JJ, editor. The pigmentary system: physiology and pathophysiology. 2 nd ed. United States: Blackwell Science; 2006. p. 780-808.
[Google Scholar]
|
| 8. |
Chang MW. Disorders of Hyperpigmentation. In: Rapini Ronald P, Bolognia Jean L, Jorizzo, Joseph L, editors. Dermatology. 2 nd ed. St. Louis: Mosby; 2008. p. 954-62.
[Google Scholar]
|
| 9. |
Shen Z, Chen L, Ye Q, Hao F, Vitorino Mdos S, Yang X, et al. Coexistent Dowling-Degos disease and reticulate acropigmentation of kitamura with progressive seborrheic keratosis. Cutis 2011;87:73-5.
[Google Scholar]
|
| 10. |
Thami GP, Jaswal R, Kanwar AJ, Radotra BD, Singh IP. Overlap of reticulate acropigmentation of Kitamura, acropigmentation of Dohi and Dowling-Degos disease in four generations. Dermatology 1998;196:350-1.
[Google Scholar]
|
| 11. |
Helbig I, Fölster-Holst R, Brasch J, Hausser I, van Baalen A, Muhle H, et al. Dyschromatosis ptychotropica: An unusual pigmentary disorder in a boy with epileptic encephalopathy and progressive atrophy of the central nervous system-a novel entity? Eur J Pediatr 2010;169:495-500.
[Google Scholar]
|
| 12. |
Jaeckle Santos LJ, Xing C, Barnes RB, Ades LC, Megarbane A, Vidal C, et al. Refined mapping of X-linked reticulate pigmentary disorder and sequencing of candidate genes. Hum Genet 2008;123:469-76.
[Google Scholar]
|
| 13. |
Urabe K, Hori Y. Dyschromatosis. Semin Cutan Med Surg 1997;16:81-5.
[Google Scholar]
|
| 14. |
Schnur RE, Heymann WR. Reticulate hyperpigmentation. Semin Cutan Med Surg 1997;16:72-80.
[Google Scholar]
|
| 15. |
Kocatürk E, Kavala M, Zindanci I, Zemheri E, Koç MK, Sarigül S. Reticulate acropigmentation of Kitamura: Report of a familial case. Dermatol Online J 2008;14:7.
[Google Scholar]
|
| 16. |
Singal A, Bhattacharya SN, Baruah MC, Indrayan A, Singh N. Is the heredity of reticulate acro pigmentation of Kitamura always autosomal dominant? J Dermatol 1998;25:57-9.
[Google Scholar]
|
| 17. |
Kanwar AJ, Kaur S, Rajagopalan M. Reticulate acropigmentation of Kitamura. Int J Dermatol 1990;29:217-9.
[Google Scholar]
|
| 18. |
Lestringant GG, Masouyé I, Frossard PM, Adeghate E, Galadari IH. Co-existence of leukoderma with features of Dowling-Degos disease: reticulate acropigmentation of Kitamura spectrum in five unrelated patients. Dermatology 1997;195:337-43.
[Google Scholar]
|
| 19. |
Wallis MS, Mallory SB. Reticulate acropigmentation of Kitamura with localized alopecia. J Am Acad Dermatol 1991;25:114-6.
[Google Scholar]
|
| 20. |
Kameyama K, Morita M, Sugaya K, Nishiyama S, Hearing VJ. Treatment of reticulate acropigmentation of Kitamura with azeleic acid. An immunohistochemical and electron microscopic study. J Am Acad Dermatol 1992;26:817-20.
[Google Scholar]
|
| 21. |
Kim NI, Park SY, Youn J. Dyschromatosis symmetrica hereditaria affecting two families. Korean J Dermatol 1980;18:585-9.
[Google Scholar]
|
| 22. |
Li M, Yang LJ, Zhu XH, Zhang HP, Dai XY. Analysis on the mutation of ADAR gene in a pedigree with dyschromatosis symmetrical hereditaria. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2007;24:446-8.
[Google Scholar]
|
| 23. |
Taki T, Kozuka S, Izawa Y, Hiramatsu M, Usuda T, Yokoo K, et al. Surgical treatment of speckled skin caused by dyschromatosis symmetrica hereditaria- a case report. J Dermatol 1986;13:471-3.
[Google Scholar]
|
| 24. |
Suzuki N, Suzuki T, Inagaki K, Ito S, Kono M, Fukai K, et al. Mutation analysis of the ADAR1 gene in dyschromatosissymmetrica hereditaria and genetic differentiation from both dyschromatosis universalis hereditaria and acropigmentatio reticularis. J Invest Dermatol 2005;124:1186-92.
[Google Scholar]
|
| 25. |
Rycroft RJ, Calnan CD, Wells RS. Universal dyschromatosis, small stature and high-tone deafness. Clin Exp Dermatol 1977;2:45-8.
[Google Scholar]
|
| 26. |
Nogita T, Mitsuhashi Y, Takeo C, Tsuboi R. Removal of facial and labial lentigines in dyschromatosis universalis hereditaria with a Q-switched alexandrite laser. J Am Acad Dermatol 2011;65:61-3.
[Google Scholar]
|
| 27. |
Hara M, Kumasaka K, Tomita Y, Tagami H. Unilateral dermatomal pigmentary dermatosis: A variant dyschromatosis? J Am Acad Dermatol 1992;27:763-4.
[Google Scholar]
|
| 28. |
Dereure O. Naegeli-Franceschetti-Jadassohn syndrome and dermatopathia pigmentosa reticularis. Two allelic ectodermal dysplasias related to mutations of dominant gene coding for keratin 14. Ann Dermatol Venereol 2007;134:595.
[Google Scholar]
|
| 29. |
Arthur EA, Brod BA, Kantor GR. Palmoplantar keratoderma associated with dermatopathia pigmentosa reticularis: Successful treatment with etretinate. Int J Dermatol 1995;34:645-6.
[Google Scholar]
|
| 30. |
Liao H, Zhao Y, Baty DU, McGrath JA, Mellerio JE, McLean WH. A heterozygous frameshift mutation in the V1 domain of keratin 5 in a family with Dowling-Degos disease. J Invest Dermatol 2007;127:298-300.
[Google Scholar]
|
| 31. |
Li M, Hunt MJ, Commens CA. Hidradenitis suppurativa, Dowling Degos disease and perianal squamous cell carcinoma. Australas J Dermatol 1997;38:209-11.
[Google Scholar]
|
| 32. |
Fenske NA, Groover CE, Lober CW, Espinoza CG. Dowling-Degos disease, hidradenitis suppurativa, and multiple keratoacanthomas. A disorder that may be caused by a single underlying defect in pilosebaceous epithelial proliferation. J Am Acad Dermatol 1991;24:888-92.
[Google Scholar]
|
| 33. |
Kim YC, Davis MD, Schanbacher CF, Su WP. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): A clinical and histopathologic study of 6 cases. J Am Acad Dermatol 1999;40:462-7.
[Google Scholar]
|
| 34. |
Wenzel G, Petrow W, Tappe K. Treatment of Dowling-Degos disease with Er:YAG-laser: Results after 2.5 years. Dermatol Surg 2003;29:1161-2.
[Google Scholar]
|
| 35. |
Wenzel G, Petrow W, Tappe K, Gerdsen R, Uerlich WP, Bieber T. Changing a concept-controversy on the confusing spectrum of the reticulate pigmented disorders of the skin. J Cutan Pathol 2009;36:44-8.
[Google Scholar]
|
| 36. |
Hanneken S, Rütten A, Pasternack SM, Eigelshoven S, El Shabrawi-Caelen L, Wenzel J, et al. Systematic mutation screening of KRT5 supports the hypothesis that Galli-Galli disease is a variant of Dowling-Degos disease. Br J Dermatol 2010;163:197-200.
[Google Scholar]
|
| 37. |
Hori Y, Kubota Y. Pigmentatio reticularis faciei et colli with multiple epithelial cysts. Arch Dermatol 1985;121:109-11.
[Google Scholar]
|
| 38. |
Sirinavin C, Trowbridge AA. Dyskeratosis congenita: Clinical features and genetic aspects. J Med Genet 1975;12:339-54.
[Google Scholar]
|
| 39. |
Tchou PK, Kohn T. Dyskeratosis congenita: An autosomal dominant disorder. J Am Acad Dermatol 1982;6:1034-9.
[Google Scholar]
|
| 40. |
Juneja HS, Elder FF, Gardner FH. Abnormality of platelet size and T-lymphocyte proliferation in an autosomal recessive form of dyskeratosis congenita. Eur J Haematol 1987;39:306-10.
[Google Scholar]
|
| 41. |
Parry EM, Alder JK, Lee SS, Phillips JA 3 rd , Loyd JE, Duggal P, et al. Decreased dyskerin levels as a mechanism of telomere shortening in X-linked dyskeratosis congenita. J Med Genet 2011;48:327-33.
[Google Scholar]
|
| 42. |
Akaboshi S, Yoshimura M, Hara T, Kageyama H, Nishikwa K, Kawakami T, et al. A case of Høyeraal-Hreidarsson syndrome: Delayed myelination and hypoplasia of corpus callosum are other important signs. Neuropediatrics 2000;31:141-4.
[Google Scholar]
|
| 43. |
Joenje H, Oostra AB, Wijker M, di Summa FM, van Berkel CG, Rooimans MA, et al. Evidence for at least eight Fanconi anemia genes. Am J Hum Genet 1997;61:940-4.
[Google Scholar]
|
| 44. |
De Kerviler E, Guermazi A, Zagdanski AM, Gluckman E, Frija J. The clinical and radiological features of Fanconi's anaemia. Clin Radiol 2000;55:340-5.
[Google Scholar]
|
| 45. |
Gougerot H, Carteaud A. Papillomatose pigmente´e innomine´e. Bull Soc Fr Dermatol Syphiligr 1927;34:719.
[Google Scholar]
|
| 46. |
Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): A minocycline-responsive dermatosis without evidence for yeast in pathogenesis: A study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol 2006;154:287-93.
[Google Scholar]
|
| 47. |
Puig L, demoragas JM. Confluent and reticulated papillomatosis of Gougerot and Carteaud: Minocycline deserves trial before etretinate. Arch Dermatol 1995;131:109-10.
[Google Scholar]
|
| 48. |
Lee MP, Stiller MJ, McClain SA, Shupack JL, Cohen DE. Confluent and reticulated papillomatosis: Response to high-dose oral isotretinoin therapy and reassessment of epidemio- logic data. J Am Acad Dermatol 1994;31:327-31.
[Google Scholar]
|
| 49. |
Böer A, Misago N, Wolter M, Kiryu H, Wang XD, Ackerman AB. Prurigo pigmentosa: A distinctive inflammatory disease of the skin. Am J Dermatopathol 2003;25:117-29.
[Google Scholar]
|
| 50. |
Erbagci Z. Prurigo pigmentosa in association with Helicobacter pylori infection in a Caucasian Turkish woman. Acta Derm Venereol 2002;82:302-3.
[Google Scholar]
|
| 51. |
Cota C, Donati P, Amantea A. Prurigo pigmentosa associated with an atopic diathesis in a 13-year-old girl. Pediatr Dermatol 2007;24:277-9.
[Google Scholar]
|
| 52. |
Miyachi Y, Yoshioka A, Horio T, Imamura S, Niwa Y. Prurigo pigmentosa: A possible mechanism of action of sulfonamides. Dermatologica 1986;172:82-8.
[Google Scholar]
|
Fulltext Views
21,309
PDF downloads
3,444





![[Figure - 1]](#fig_ijdvl_2013_79_1_17_104665_f4.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2013_79_1_17_104665_t1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2013_79_1_17_104665_f5.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2013_79_1_17_104665_f6.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2013_79_1_17_104665_f7.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2013_79_1_17_104665_f8.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2013_79_1_17_104665_t2.jpg){kind=link}
![[Table - 3]](#tbl_ijdvl_2013_79_1_17_104665_t3.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2013_79_1_17_104665_f9.jpg){kind=link}
![[Figure - 7]](#fig_ijdvl_2013_79_1_17_104665_f10.jpg){kind=link}
![[Figure - 8]](#fig_ijdvl_2013_79_1_17_104665_f11.jpg){kind=link}
![[Figure - 9]](#fig_ijdvl_2013_79_1_17_104665_f12.jpg){kind=link}
![[Figure - 10]](#fig_ijdvl_2013_79_1_17_104665_f13.jpg){kind=link}
![[Figure - 11]](#fig_ijdvl_2013_79_1_17_104665_f14.jpg){kind=link}
![[Figure - 12]](#fig_ijdvl_2013_79_1_17_104665_f15.jpg){kind=link}