Translate this page into:
Familial progressive hypo- and hyperpigmentation: A variant case
2 The First Affiliated Hospital of Nanjing Medical University, Nanjing, China
Correspondence Address:
Wen-yuan Zhu
Department of Dermatology, the First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029
China
| How to cite this article: Zhang Rz, Zhu Wy. Familial progressive hypo- and hyperpigmentation: A variant case. Indian J Dermatol Venereol Leprol 2012;78:350-353 |
Abstract
Familial progressive hyper- and hypopigmentation (FPHH) is characterized by diffuse hyperpigmentation with variable intensity. Cafe'-au-lait macules and larger hypopigmented ash-leaf macules are also present. Herein, we reported a variant case of FPHH. The patient was a two-year-old Chinese girl showing diffuse hyper- and hypopigmented lesions, longitudinal melanonychia in both thumbs, and infantile seizures, without any lentigines.Introduction
Some children develop excessive pigmentation of the skin soon after birth or in the first year of life. Several authors have described this generalized hypermelanosis without systemic or developmental defects but often with a familial pattern as melanosis universalis hereditaria (MUH), melanosis diffusa congenital, universal acquired melanosis, familial progressive hyperpigmentation (FPH) (OMIM-145250) and familial universal or diffuse melanosis. It is still not clear whether these different entities represent the same disorder or they are distinct. The pathogenesis of FPH is not understood to date. Two loci have been identified for FPH: One on chromosome 19p13-pter and another on 12q21.31-q23.1. [1] dyschromatosis universalis hereditaria 2 (DUH2) was subsequently linked to the latter. [2]
Familial progressive hyper- and hypopigmentation (FPHH) is thought to be an autosomal dominant disorder with reduced penetrance. Clinical signs consist of progressive diffuse, partly blotchy hyperpigmented lesions, multiple cafe′-au-lait spots, intermingled with scattered hypopigmented-appearing maculae and lentigines. FPHH is distinct from FPH, in which no hypopigmented features are present, and which is phenotypically and histologically closer to DUH2. The findings observed by Amyere et al., suggest that the same gene (KITLG) is behind FPH, FPHH, and likely DUH2, and underscores the importance of the KITLG b-strand motif VTNN in the activation of c-Kit. [3] In one Chinese family with FPH without cafe′-au-lait spots, a single amino-acid change in KITLG was reported. Two FPHH families with the same substitution and two new substitutions in another two FPHH families were observed. Herein, we report one case that seems to best resemble those patients diagnosed with FPHH, but has other features also.
Case Report
A two-year-old Chinese girl presented to our dermatology department with uniform hyperpigmentation with numerous imposed hyper- and hypopigmented macules, which had been noted since birth and had become more noticeable with age. The child was born to healthy nonconsanguineous parents, following an uneventful pregnancy. The girl had mild mental retardation and epilepsy which happened frequently during the first year. The longest persistence time of a generalized seizures exceeded 10 h. The epileptiform seizure was always accompanied by hyperthermia, frequently over 40°C. Anticonvulsant therapy has been used for controlling this seizure for one year. She had a history of repeated infections. History provides no evidence of chronic chemical poisoning. The patient belongs to yellow race, has neither negroid ancestors nor negroid characteristics. The family history revealed that the maternal grandmother, great grandmother and an uncle-in-law had diffuse hyperpigmented skin. However, the infant′s mother was free of skin lesions. The girl′s father and his family have no history of skin or neurologic lesions. The pedigree of the patient was as follows [Figure - 1].
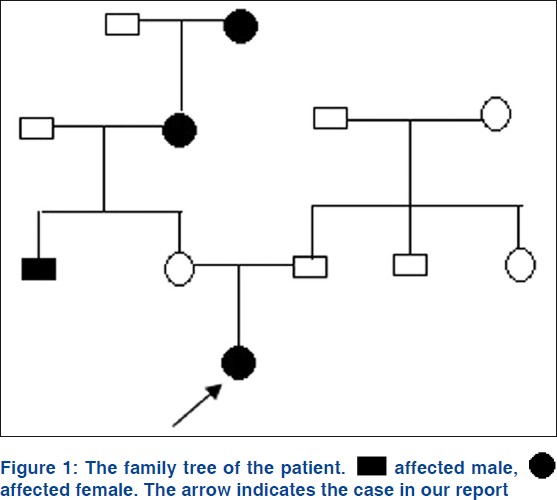 |
| Figure 1: |
Physical examination revealed numerous pigmented spots on her face, which were 2-3 mm in diameter, with ill-defined borders and formed a mosaic pattern giving the skin a ′dirty′ appearance [Figure - 2]. The girl′s trunk was almost uniformly deep bronze-brown in color with some superimposed irregular café -au-lait spots, in the sizes of 0.5-5 cm, together with 4 ash-leaf-like white macules [Figure - 3]a-d. Skin over the joints (ankle, knee, elbow, wrist joints and knuckles), axillae, neck, nipples and fovea umbilicalis was markedly hyperpigmented. Longitudinal melanonychia (multiple bands) was also observed in her both thumbs [Figure - 3]e and f. The proximal part of all fingers′ nail beds were slightly pigmented. Several brown maculae were seen over buccal mucosa and on the gums. No generalized lentiginosis, axillary freckling, abnormal hairs and teeth were observed. The girl′s mother refused our request for biopsies of these lesions.
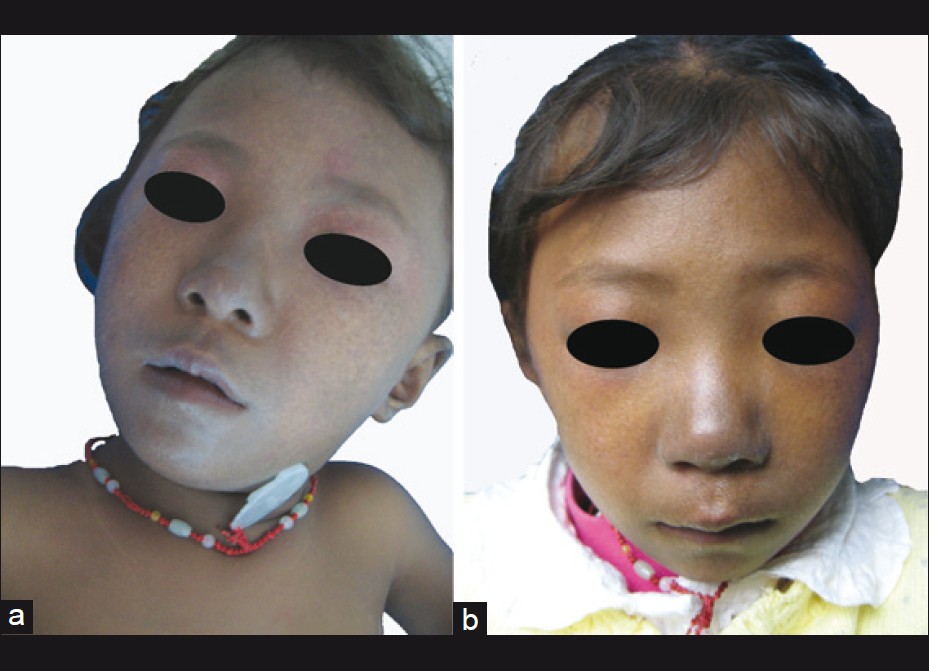 |
| Figure 2: Reticulate network of hyperpigmented macules over the face. (a) At the initial presentation; (b) 10 months later |
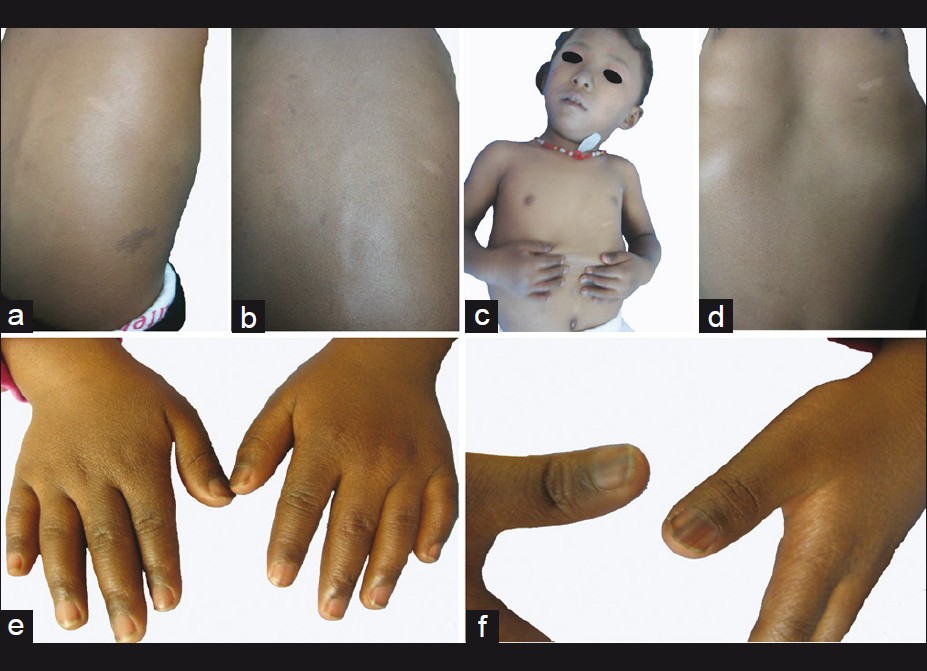 |
| Figure 3: Some irregular café au - lait spots and Ash - leaf - like white macules were superimposed on the uniformly deep bronze-brown skin. (a and b) dorsal view; (c and d) anterior view. (e) markedly hyperpigmented skin over the knuckles; (f) longitudinal melanonychia (multiple bands) in her both thumbs |
The patient had no evidence of pituitary-adrenal pathology, nor were β-MSH levels found to be increased. Routine laboratory tests, including urinary and plasma aminograms, determinations of 24 h urinary porphyrins, stool examination for ova and parasites, X-ray examinations (chest, skull, thoracic spine, mandible, hands, upper-gastrointestinal tract, small bowel and colon), several times repeated were within the limits of normal. Electroencephalography (EEG) exhibited slight abnormality. There was no electrocardiographic abnormality. Computed tomography (CT) of brain revealed no significant parenchymal abnormalities. Further screening, including neurological, ophthalmological and orthopaedic investigations did not reveal any abnormalities. The patient was followed up in our department for periodic general evaluation of her skin. After a follow up of 10 months, comparing the photographs taken in 2010, her whole skin became lighter, whereas the preexistent hyperpigmentation and hypopigmentation areas had remained unaltered. Based on the clinical features and her family history, the diagnosis of FPHH was made.
Discussion
In the English literatures, we found five papers dealing with familial hyperpigmentation and hypopigmentation. Westerhof et al., described the concurrent presence of hyper- and hypopigmented patches in a three-generation family with the affected family members also showing growth retardation and mental deficiency. [4] Hoo et al., reported on an unusual genetic condition in a three generation family of French-Canadian ancestry with a concurrent presence of cafe-au lait and hypopigmented macules/patches. [5] Furthermore, the lesion pattern changed with progressive age, from circumscribed and large cafe-au-lait and hypopigmented macules/patches to hyperpigmented and hypopigmented freckling. In 2004, Zanardo et al., reported on five patients from three different families presenting with a peculiar progressive pigmentary disorder. [6] All families are clustered in a small area around the town of Teublitz in south-east Germany. Pedigree analysis is compatible with an autosomal dominant mode of inheritance with variable penetrance. These patients showed a progressive diffuse, partly blotchy, hyperpigmentation, intermixed with scattered small hypopigmented maculae, a few large hypopigmented areas, occasional cafe′-au-lait spots and, most remarkably, a generalized lentiginosis. The authors presumed that this condition represented a rare and perhaps novel variant of FPH. In 2011, Amyere et al., first introduced the term of FPHH. Seven families (including a forementioned three families) with this pigmentary disorder were studied. [3] Whole-genome scan was performed on all available family members, and linkage to 12q21.12-q22, and a KITLG mutation in four families were identified by multipoint linkage analyses. The authors believed that FPHH is distinct from FPH, in which no hypopigmented features are present.
Several human genetic diseases, such as DUH, dyschromatosis symmetrica hereditaria (DSH), and xeroderma pigmentosum (XP), show some overlap with FPH and FPHH. DUH is characterized by sharply demarcated brown macules of various sizes on diffusely hypopigmented skin, typically involving the head, anterior chest, abdomen and extremities mostly without involvement of palms and soles. However, cafe′-au-lait macules and larger hypopigmented ash-leaf macules are not typical features of this condition, making it possible to distinguish such patients from ours. DSH shows a characteristic mixture of hyperpigmented and hypopigmented macules of various sizes, limited largely to the dorsal aspects of the hands and feet. In addition, the genes for DSH and DUH have previously been mapped to chromosomes 1q21.3, 6q24.2-q25.2, and 12q21-q23. Thus, the genetic basis of FPH (12q21.31-q23.1) and FPHH (12q21.12-q22) is likely to differ from that of DSH and DUH. [7] Interestingly, the p.Asn36Ser amino-acid change causes both FPH and FPHH, suggesting additional factors to have a role in the development of the phenotype.
Our patient had a background of uniformly congenital hyperpigmentation, with some superimposed irregular café -au-lait spots and ash-leaf-like white macules. So, FPH and universal acquired melanosis (Carbon baby) could be excluded. [8] In view of the lack of any other clinical signs, the differential diagnosis from other diseases or syndromes characterized by an unusual lentiginous pattern of hyperpigmentation, such as LEOPARD syndrome, tuberous sclerosis, NAME syndrome or Peutz-Jeghers syndrome, was easy, as all these syndromes show different additional abnormalities in other organs. [9]
Comparing with the previous report, our case is of interest for four reasons. Firstly, the diffuse and uniformly hyperpigmented background, appearing uniformly deep bronze-brown skin, gave us deep impression and made us think of FPH or Carbon baby. Especially, the girl′s face presented dirty appearance of reticulate pigmentation, which has not previously been traced in the literature. Secondly, longitudinal melanonychia (multiple bands) was also observed in her both thumbs at the birth. This condition may imply an abnormal development of melanoblasts in the nail beds. Thirdly, the girl had mild mental retardation, epileptic seizure that happened frequently during the first year, impaired temperature regulation (hyperthemia) and increased susceptibility to infection. In conclusion, the genetic determination of pigmentation is a complex process gradually becoming understood by the identification of genes causing familial phenotypes. Unfortunately, contact with the patient has been lost, and blood samples for genetic marker studies are not available now.
| 1. |
Wang ZQ, Si L, Tang Q, Lin D, Fu Z, Zhang J, et al. Gain-of-function mutation of KIT ligand on melanin synthesis causes familial progressive hyperpigmentation. Am J Hum Genet 2009;84:672-7.
[Google Scholar]
|
| 2. |
Stuhrmann M, Hennies HC, Bukhari IA, Brakensiek K, Nürnberg G, Becker C, et al. Dyschromatosis universalis hereditaria: Evidence for autosomal recessive inheritance and identification of a new locus on chromosome 12q21-q23. Clin Genet 2008;73:566-72.
[Google Scholar]
|
| 3. |
Amyere M, Vogt T, Hoo J, Brandrup F, Bygum A, Boon L, et al. KITLG mutations cause familial progressive hyper- and hypopigmentation. J Invest Dermatol 2011;131:1234-9.
[Google Scholar]
|
| 4. |
Westerhof W, Beemer FA, Cormane RH, Delleman JW, Faber WR, de Jong JG, et al. Hereditary congenital hypopigmented and hyperpigmented macules. Arch Dermatol 1978;114:931-6.
[Google Scholar]
|
| 5. |
Hoo JJ, Shrimpton AE. Familial hyper- and hypopigmentation with age-related pattern change. Am J Med Genet 2005;132A:215-8.
[Google Scholar]
|
| 6. |
Zanardo L, Stolz W, Schmitz G, Kaminski W, Vikkula M, Landthaler M, et al. Progressive hyperpigmentation and generalized lentiginosis without associated systemic symptoms: A rare hereditary pigmentation disorder in south-east Germany. Acta Derm Venereol 2004;84:57-60.
[Google Scholar]
|
| 7. |
Dereure O. Progressive familial hyperpigmentation is associated with mutations activating the KIT receptor ligand. Ann Dermatol Venereol 2010;137:247.
[Google Scholar]
|
| 8. |
Kaviarasan PK, Prasad PV, Joe JM, Nandana N, Viswanathan P. Universal acquired melanosis (Carbon baby). Indian J Dermatol Venereol Leprol 2008;74:38-40.
[Google Scholar]
|
| 9. |
Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S, et al. Peutz-Jeghers syndrome: A systematic review and recommendations for management. Gut 2010;59:975-86.
[Google Scholar]
|
Fulltext Views
3,206
PDF downloads
1,038





![[Figure - 1]](#fig_ijdvl_2012_78_3_350_95453_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_3_350_95453_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2012_78_3_350_95453_f3.jpg){kind=link}